


![]()
|
|
![]()

Documenti
Legislazione
Direttive Sicurezza
Timeline Sicurezza IT
D.Lgs. 81/2008
Interpelli
Soggetti abilitati
ATECO / Livello rischio
Cassazione
Convenzioni ILO
TUSSL / Link


Documenti
Legislazione
Testo Unico Ambiente
Interpelli ambientali
Elenco CER
RENTRi


Documenti
Legislazione
CLP consolidato
REACH Consolidato
Seveso III
PXXX | EUH | HXXX
Elenco gas tossici
Parametri acque

Documenti
Legislazione
Norme armonizzate Click
Direttiva click
Nuovo approccio


Attività PI
D.P.R. 151/2011
Moduli PI
Codice RTO II
Norme PI 2022
Decreti PI Sett. 2021


Documenti
Legislazione
Tremcards
Etichette ADR
Kemler

Regolamento macchine
Direttiva macchine
Documenti Regolamento
Norme armonizzate
CEM4 | Software

HACCP (CE) 852/2004
MOCA (CE) 1935/2004
GMP (CE) 2023/2006
MOCA - GMP consolidato
Food Safety book

Documenti
Legislazione
DM. n. 37/2008
Legge n. 10/1991

Norme armonizzate
Direttiva click
Prassi di riferimento








ID 2047 | Update Rev. 35.0 2024 (06.07.2024) - In aggiornamento
Allegate tutte le Guide ufficiali sui i Dispositivi medici in accordo con la Direttiva 93/42/CEE e il nuovo Regolamento (UE) 2017/745.
Download 0. Raccolta linee guida MEDDEV e MDCG Indice 26.02.2024
_______
New Rev. 35.0 del 06 Luglio 2024:
- MDCG 2024-10 Clinical evaluation of orphan medical devices June 2024
________
Regolamento (UE) 2017/745 concernente i dispositivi medici (si applica dal 20 Maggio 2021)
[box-download]In calce all'articolo pdf "List of Guidance MEDDEVs e MDCG Rev. 33.0 Update 28.09.2023" riservato Abbonati[/box-download]
New regulations
Guidance documents to assist stakeholders in implementing the Medical Devices Regulations.
1. MDCG Documents
MDCG endorsed documents
| Reference | Title | Publication date |
| MDCG 2024-10 | Clinical evaluation of orphan medical devices | June 2024 |
| MDCG 2024-2 | Procedures for the updates of the EMDN | February 2024 |
| MDCG 2024-1-4 | DSVG 04 on Breast implants 4 | January 2024 |
| MDCG 2024-1-3 | DSVG 03 on Cardiac implantable electronic devices (CIEDs) | January 2024 |
| MDCG 2024-1-2 | DSVG 02 on Coronary stents | January 2024 |
| MDCG 2024-1-1 | DSVG 01 on Cardiac ablation | January 2024 |
| MDCG 2024-1 | Device Specific Vigilance Guidance (DSVG) Template | January 2024 |
| MDCG 2023-7 | Guidance on exemptions from the requirements to perform clinical investigations pursuant to Article 61(4)-(6) MDR and on ‘sufficient levels of access’ to data needed to justify claims of equivalence | December 2023 |
| MDCG 2023-6 | Guidance on demonstration of equivalence for Annex XVI products - A guide for manufacturers and notified bodies | December 2023 |
| MDCG 2023-5 | Guidance on qualification and classification of Annex XVI products - A guide for manufacturers and notified bodies | December 2023 |
| MDCG 2022-11 - Rev.1 | MDCG Position Paper: Notice to manufacturers to ensure timely compliance with MDR and IVDR requirements | November 2023 |
| MDCG 2021-27 - Rev.1 | Questions and Answers on Articles 13 & 14 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746 | December 2023 |
| MDCG 2021-6 - Rev.1 | Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation | December 2023 |
| MDCG 2019-7 - Rev.1 | Guidance on article 15 of the medical device regulation (MDR) and in vitro diagnostic device regulation (IVDR) on a ‘person responsible for regulatory compliance’ (PRRC) | December 2023 |
| MDCG 2023-3 | Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices | February 2023 |
| MDCG 2023-2 MDR form List of Standard |
List of Standard Fees | January 2023 |
| MDCG 2023-2 IVDR form List of Standard |
List of Standard Fees | January 2023 |
| MDCG 2023-2 | List of Standard Fees | January 2023 |
| MDCG 2023-1 | Guidance on the health institution exemption under Article 5(5) of Regulation (EU) 2017/745 and Regulation (EU) 2017/746 | January 2023 |
| MDCG 2022- 21 | Guidance on Periodic Safety Update Report (PSUR) according to Regulation (EU) 2017/745 | December 2022 |
| MDCG 2022- 20 | Substantial modification of performance study under Regulation (EU) 2017/746 | December 2022 |
| MDCG 2022-19 | Performance study application/notification documents under Regulation (EU) 2017/746 | December 2022 |
| Q&A Rev. 1 | Q&A on practical aspects related to the implementation of Regulation (EU) 2023/607 - Extension of the MDR transitional period and removal of the “sell off” periods | July 2023 |
| MDCG 2022-18 ADD.1 | MDCG Position Paper on the application of Article 97 MDR to legacy devices for which the MDD or AIMDD certificate expires before the issuance of a MDR certificate - Addendum 1 | June 2023 |
| MDCG 2022-18 | MDCG Position Paper on the application of Article 97 MDR to legacy devices for which the MDD or AIMDD certificate expires before the issuance of a MDR certificate | December 2022 |
| MDCG 2022-17 | MDCG position paper on "hybrid audits" | December 2022 |
| MDCG 2022-16 | Guidance on Authorised Representatives Regulation (EU) 2017/745 and Regulation (EU) 2017/746 | October 2022 |
| MDCG 2022-15 | Guidance on appropriate surveillance regarding the transitional provisions under Article 110 of the IVDR with regard to devices covered by certificates according to the IVDD | September 2022 |
| MDCG 2022-14 | Transition to the MDR and IVDR - Notified body capacity and availability of medical devices and IVDs | August 2022 |
| MDCG 2022-13 | Designation, re-assessment and notification of conformity assessment bodies and notified bodies | August 2022 |
| MDCG 2022-12 | Guidance on harmonised administrative practices and alternative technical solutions until Eudamed is fully functional (for Regulation (EU) 2017/746 on in vitro diagnostic medical devices) | July 2022 |
| MDCG 2022-11 | MDCG Position Paper: Notice to manufacturers to ensure timely compliance with MDR requirements | May 2022 |
| MDCG 2022-10 | Q&A on the interface between Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) | May 2022 |
| MDCG 2022-9 | Summary of safety and performance template | May 2022 |
| MDCG 2022-8 | Regulation (EU) 2017/746 - application of IVDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2022 in accordance with Directive 98/79/EC | May 2022 |
| MDCG 2022-7 | Q&A on the Unique Device Identification system under Regulation (EU) 2017/745 and Regulation (EU) | May 2022 |
| MDCG 2022-6 | Guidance on significant changes regarding the transitional provision under Article 110(3) of the IVDR | May 2022 |
| MDCG 2022-5 | Guidance on borderline between medical devices and medicinal products under Regulation (EU) 2017/745 on medical | May 2022 |
| MDCG 2022-4 rev.1 | Guidance on appropriate surveillance regarding the transitional provisions under Article 120 of the MDR with regard to devices covered by certificates according to the MDD or the AIMDD | December 2022 |
| MDCG 2022-4 | Guidance on appropriate surveillance regarding the transitional provisions under Article 120 of the MDR | February 2022 |
| MDCG 2022-3 | Verification of manufactured class D IVDs by notified bodies | February 2022 |
| MDCG 2022-2 | Guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices (IVDs) | January 2022 |
| MDCG 2022-1 | Notice to 3rd country manufacturers of SARS-CoV-2 in vitro diagnostic medical devices | January 2022 |
| MDCG 2021-28 | Substantial modification of clinical investigation under Medical Device Regulation | December 2021 |
| MDCG 2021-27 | Questions and Answers on Articles 13 & 14 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746 | December 2021 |
| MDCG 2021-26 | Questions and Answers on repackaging & relabelling activities under Article 16 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746 | October 2021 |
| MDCG 2021-25 | Regulation (EU) 2017/745 - application of MDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2021 in accordance with Directives 90/385/EEC or 93/42/EEC | October 2021 |
| MDCG 2021-24 | Guidance on classification of medical devices | October 2021 |
| Helsinki Procedure | Helsinki Procedure for borderline and classification under MDR & IVDR | September 2021 |
| MDCG 2021-23 | Guidance for notified bodies, distributors and importers onClarification on “first certification for that type of device” and corresponding procedures to be followed by notified bodies, in context of the consultation of the expert panel referred to in Article 48(6) of Regulation (EU) 2017/746 | August 2021 |
| MDCG 2021-22 | Clarification on “first certification for that type of device” and corresponding procedures to be followed by notified bodies, in context of the consultation of the expert panel referred to in Article 48(6) of Regulation (EU) 2017/746 | August 2021 |
| MDCG 2021-21 | Guidance on performance evaluation of SARS-CoV-2 in vitro diagnostic medical devices | August 2021 |
| MDCG 2021-20 | Instructions for generating CIV-ID for MDR Clinical Investigations | July 2021 |
| MDCG 2021-19 | Guidance note integration of the UDI within an organisation’s quality management system | July 2021 |
| MDCG 2021-18 | Applied-for scope of designation and notification of a conformity assessment body – Regulation (EU) 2017/746 (IVDR) | July 2021 |
| MDCG 2021-17 | Applied-for scope of designation and notification of a conformity assessment body – Regulation (EU) 2017/745 (MDR) | July 2021 |
| MDCG 2021-16 | Application form to be submitted by a conformity assessment body when applying for designation as notified body under the in vitro diagnostic devices regulation (IVDR) | July 2021 |
| MDCG 2021-15 | Application form to be submitted by a conformity assessment body when applying for designation as notified body under the medical devices regulation (MDR) | July 2021 |
| MDCG 2021-14 | Explanatory note on IVDR codes | July 2021 |
| MDCG 2021-13 rev. 1.0 | Questions and answers on obligations and related rules for the registration in EUDAMED of actors other than manufacturers, authorised representatives and importers subject to the obligations of Article 31 MDR and Article 28 IVDR | July 2021 |
| MDCG 2021-12 | FAQ on the European Medical Device Nomenclature (EMDN) | May 2021 |
| MDCG 2021-11 | Guidance on Implant Card – Device types | May 2021 |
| MDCG 2021-10 | The status of Appendixes E-I of IMDRF N48 under the EU regulatory framework for medical devices | May 2021 |
| MDCG 2021-9 | MDCG Position Paper on the Implementation of UDI requirements for contact lenses, spectacle frames, spectacle lenses & ready readers | May 2021 |
| MDCG 2021-8 | Clinical investigation application/notification documents | May 2021 |
| MDCG 2021-7 | Notice to manufacturers and authorised representatives on the impact of genetic variants on SARS-COV-2 in vitro diagnostic medical devices | May 2021 |
| MDCG 2021-6 | Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation | May 2021 |
| MDCG 2021-5 | Guidance on standardisation for medical devices | April 2021 |
| MDCG 2021-4 | Application of transitional provisions for certification of class D in vitro diagnostic medical devices according to Regulation (EU) 2017/746 | April 2021 |
| MDCG 2021-3 | Questions and Answers on Custom-Made Devices | March 2021 |
| MDCG 2021-2 | Guidance on state of the art of COVID-19 rapid antibody tests | March 2021 |
| MDCG 2021-1 | Guidance on harmonised administrative practices and alternative technical solutions until EUDAMED is fully functional | Febraury 2021 |
| MDCG 2020-18 | MDCG Position Paper on UDI assignment for Spectacle lenses & Ready readers | December 2020 |
| MDCG 2020-17 | Questions and Answers related to MDCG 2020-4: “Guidance on temporary extraordinary measures related to medical device notified body audits during COVID-19 quarantine orders and travel restrictions” |
December 2020 |
|
MDCG 2020-16 Rev.2 |
Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746 | Febraury 2023 |
| MDCG 2020-16 | Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746 | November 2020 |
| MDCG 2020-15 | MDCG Position Paper on the use of the EUDAMED actor registration module and of the Single Registration Number (SRN) in the Member States | August 2020 |
| MDCG 2020-14 | Guidance for notified bodies on the use of MDSAP audit reports in the context of surveillance audits carried out under the Medical Devices Regulation (MDR)/In Vitro Diagnostic medical devices Regulation (IVDR) | August 2020 |
| MDCG 2020-13 | Clinical evaluation assessment report template | July 2020 |
| MDCG 2020-12 | Guidance on transitional provisions for consultations of authorities on devices incorporating a substance which may be considered a medicinal product and which has action ancillary to that of the device, as well as on devices manufactured using TSE susceptible animal tissues | June 2020 |
| MDCG 2020-11 | Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745 | May 2020 |
| MDCG 2020-10-2 | Clinical Investigation Summary Safety Report Form v1.0 | May 2020 |
| MDCG 2020-10-1 | Guidance on the renewal of designation and monitoring of notified bodies under Directives 90/385/EEC and 93/42/EEC to be performed in accordance with Commission Implementing Regulation (EU) 2020/666 amending Commission Implementing Regulation | May 2020 |
| MDCG 2020-9 | Regulatory requirements for ventilators and related accessories | April 2020 |
| MDCG 2020-8 | Post-market clinical follow-up (PMCF) Evaluation Report Template | April 2020 |
| MDCG 2020-7 | Post-market clinical follow-up (PMCF) Plan Template | April 2020 |
| MDCG 2020-6 | Regulation (EU) 2017/745: Clinical evidence needed for medical devices previously CE marked under Directives 93/42/EEC or 90/385/EEC | April 2020 |
| MDCG 2020-5 | Clinical Evaluation - Equivalence | April 2020 |
| MDCG 2020-4 | Guidance on temporary extraordinary measures related to medical device Notified Body audits during COVID-19 quarantine orders and travel restrictions | April 2020 |
| MDCG 2020-3 Rev.1 | Guidance on significant changes regarding the transitional provision under Article 120 of the MDR with regard to devices covered by certificates according to MDD or AIMDD | September 2023 |
| MDCG 2020-3 | Guidance on significant changes regarding the transitional provision under Article 120 of the MDR with regard to devices covered by certificates according to MDD or AIMDD | March 2020 |
| MDCG 2020-2 | Class I Transitional provisions under Article 120 (3 and 4) – (MDR) | March 2020 |
| MDCG 2020-1 | Guidance on Clinical Evaluation (MDR) / Performance Evaluation (IVDR) of Medical Device Software | March 2020 |
| MDCG 2019-16 | Guidance on Cybersecurity for medical devices | December 2019 |
| MDCG 2019-15 | Guidance notes for manufacturers of class I medical devices | December 2019 |
| MDCG 2019-14 | Explanatory note on MDR codes | December 2019 |
| MDCG 2019-13 | MDCG 2019-13 Guidance on sampling of MDR Class IIa / Class IIb and IVDR Class B / Class C devices for the assessment of the technical documentation | December 2019 |
| MDCG 2019-12 | Designating authority's final assessment form: Key information (EN) | October 2019 |
| MDCG 2019-11 |
Qualification and classification of software - Regulation (EU) 2017/745 and Regulation (EU) 2017/746 |
October 2019 |
| MDCG 2019-10 |
Application of transitional provisions concerning validity of certificates issued in accordance to Directives 90/385/EEC and 93/42/EEC |
October 2019 |
| MDCG 2019-9 |
MDCG 2019-9 Summary of safety and clinical performance A guide for manufacturers and notified bodies |
September 2019 |
| MDCG 2019-8 v2 |
Implant Card relating to the application of Article 18 Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices |
March 2020 |
| MDCG 2019-8 | Medical Devices: Guidance document Implant Card relating to the application of Article 18 Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices | July 2019 |
| MDCG 2019-7 | Guidance on Article 15 of the Medical Device Regulation (MDR) and in vitro Diagnostic Device Regulation (IVDR) regarding a ‘person responsible for regulatory compliance’ (PRRC) | June 2019 |
| MDCG 2019-6 v3 | Questions and answers: Requirements relating to notified bodies | October 2021 |
| MDCG 2019-5 | Registration of legacy devices in EUDAMED | April 2019 |
| MDCG 2019-4 | Timelines for registration of device data elements in EUDAMED | April 2019 |
| MDCG 2019-3 | Interpretation of Article 54(2)b Rev.1 | April 2020 |
| MDCG 2019-2 | Guidance on application of UDI rules to device-part of products referred to in Article 1(8), 1(9) and 1(10) of Regulation 745/2017 | Febraury 2019 |
| MDCG 2019-1 | MDCG guiding principles for issuing entities rules on Basic UDI-DI | January 2019 |
| MDCG 2018-1 v3 | Guidance on BASIC UDI-DI and changes to UDI-DI | March 2020 |
| MDCG 2018-1 | Draft guidance on basic UDI-DI and changes to UDI-DI | March 2018 |
| MDCG 2018-2 | Future EU medical device nomenclature – Description of requirements | March 2018 |
| MDCG 2018-3 | Guidance on UDI for systems and procedure packs | October 2018 |
| MDCG 2018-4 | Annex: UDI database Definitions/Descriptions and formats of the UDI core elements for systems or procedure packs | October 2018 |
| MDCG 2018-5 | UDI Assignment to Medical Device Software | October 2018 |
| MDCG 2018-6 | Clarifications of UDI related responsibilities in relation to Article 16 of the Medical Device Regulation (EU) 2017/745 and the In-Vitro Diagnostic Medical Device Regulation (EU) 2017/746 | October 2018 |
| MDCG 2018-7 | Provisional considerations regarding language issues associated with the UDI database (Annex VI, Part A Section 2 and Part B of the Medical Device Regulation (EU) 2017/745 (MDR) and the In-Vitro Diagnostic Medical Device Regulation (EU) 2017/746 (IVDR)) | October 2018 |
| MDCG 2018-8 | Guidance on content of the certificates, voluntary certificate transfers | November 2018 |
Designation of notified bodies under the new Regulations on medical devices
| Notified BODIES | Designation of notified bodies under the new Regulations on medical devices |
|---|---|
| 1. Best practice guidance on designation and notification of conformity assessment bodies (NBOG BPG 2017-1) | |
| 2. Best practice guidance on the information required for conformity assessment bodies' personnel involved in conformity assessment activities (NBOG BPG 2017-2) | |
| 3. Application form to be submitted by a conformity assessment body when applying for designation as notified body under the medical devices Regulation (MDR) (NBOG F 2017-1) | |
| 4. Application form to be submitted by a conformity assessment body when applying for designation as notified body under the in vitro diagnostic devices Regulation (IVDR) (NBOG F 2017-2) | |
| 5. Applied-for scope of designation and notification of a conformity assessment body – Regulation (EU) 2017/745 (MDR) (NBOG F 2017-3) | |
| 6. Applied-for scope of designation and notification of a conformity assessment body – Regulation (EU) 2017/746 (IVDR) (NBOG F 2017-4) | |
| 7. Preliminary assessment review template (MDR) (NBOG F 2017-5) | |
| 8. Preliminary assessment review template (IVDR) (NBOG F 2017-6) | |
| 9. Review of qualification for the authorisation of personnel (MDR) (NBOG F 2017-7) | |
| 10. Review of qualification for the authorisation of personnel (IVDR) (NBOG F 2017-8) |
Other documents
| Reference | Title | Publication date |
| UDIWG 2018-1 | UDI database. Definitions, descriptions and formats of the UDI core elements | March 2018 |
| UDIWG 2018-2 | The architecture of the UDI database - Basic UDI-DI and UDI-DI attributes for medical devices and in-vitro diagnostic medical devices | March 2018 |
Current legislation
Guidance documents to assist stakeholders in implementing directives related to medical devices.
2. MEDDEVs Guidances
The MEDDEVs promote a common approach to be followed by manufacturers and Notified Bodies that are involved in conformity assessment procedures.
- The MEDDEVs are drafted by authorities charged with safeguarding public health. This is in accordance with the relevant annexes of the Directives;
- MEDDEVs are carefully drafted through a consultation process with all interested parties and are subject to a regular updating process;
- These documents have particular reference codes and are endorsed at the Medical Devices Expert Group (MDEG) plenary meetings;
- The guidelines are not legally binding. However, due to the participation of the aforementioned interested parties and the experts from competent authorities, it is expected that the guidelines be followed, ensuring the uniform application of relevant Directive provisions.
Disclaimer: Please note that the amendments introduced by Directive 2007/47/EC or previous amending directives have not yet been incorporated into all MEDDEVs.
List of Guidance MEDDEVs
See below a complete list of all Guidance Meddevs, including links to further information:
2.12 Market surveillance
MEDDEV 2.12/1 rev.8
Guidelines on a Medical Devices Vigilance System
January 2013
Ⅰ. MEDDEV 2.12/1 rev.8 – Latest Version Forms
MEDDEV 2.12/1 rev. 7 MIR and FSCA are still valid
Active PDF forms
How to use FSCA and MIR forms
Manufacturer Incident Report - MIR Version 7.2.1
Field Safety Corrective Action - FSCA
MIR and FSCA xml files (FSCA Report - Incident Report) Version 7.2.1
Other forms and templates
Field Safety Notice Template
Trend Report
Periodic Summary Report
EU Vigilance Pilot on Trending – Additional MIR Form
EU Vigilance Pilot MIR form
EU Vigilance Pilot MIR Step-by-Step Guide
EU Vigilance Pilot Toolkit for Users
Ⅱ. Device Specific Vigilance Guidance
DSVG Template
DSVG 00 Introduction to Device Specific Vigilance Guidance
DSVG 01 Cardiac Ablation Vigilance Reporting Guidance
DSVG 02 Coronary Stents Vigilance Reporting Guidance
| Title | |
|---|---|
| 2.1 Scope, field of application, definition | MEDDEV 2.1/1 Definitions of “medical devices”, “accessory” and “manufacturer” April 1994 |
| MEDDEV 2.1/2 rev.2 Field of application of directive “active implantable medical devices” April 1994 |
|
| MEDDEV 2.1/2.1 Field of application of directive “active implantable medical devices” February 1998 |
|
| MEDDEV 2.1/3 rev.3 Borderline products, drug-delivery products and medical devices incorporating,as integral part, an ancillary medicinal substance or an ancillary human blood derivative December 2009 |
|
| MEDDEV 2.1/4 Interface with other directives – Medical devices/directive89/336/EEC relating to electromagnetic compatibility and directive 89/686/EEC relating to personal protective equipment March 1994 For the relation between the MDD and directive 89/686/EEC concerning personal protective equipment, please see the Commission services interpretative document of 21 August 2009 |
|
| MEDDEV 2.1/5 Medical devices with a measuring function June 1998 |
|
| MEDDEV 2.1/6 Qualification and Classification of stand alone software July 2016 |
|
| 2.2 Essential requirements | MEDDEV 2.2/1 rev.1 EMC requirements February 1998 |
| MEDDEV 2.2/3 rev.3 “Use by”-date June 1998 |
|
| MEDDEV 2.2/4 Conformity assessment of In Vitro Fertilisation (IVF) and Assisted Reproduction Technologies (ART) products January 2012 |
|
| 2.4 Classification MD | MEDDEV 2.4/1 rev.9 Classification of medical devices June 2010 |
| 2.5 Conformity assessment procedure |
General rules |
| Quality assurance. GHTF/SG4/N83:2010 - GHTF/SG4/N84:2010 Regulatory auditing of quality systems of medical device manufacturers (See document in the GHTF-Global Harmonization Task Force) |
|
| MEDDEV 2.5/3 rev.2 Subcontracting quality systems related June 1998 |
|
| MEDDEV 2.5/5 rev.3 Translation procedure February 1998 |
|
| MEDDEV 2.5/6 rev.1 Homogenous batches (verification of manufacturers' products) February 1998 |
|
| Conformity assessment for particular groups of products | |
| MEDDEV 2.5/7 rev.1 Conformity assessment of breast implants July 1998 |
|
| MEDDEV 2.5/9 rev.1 Evaluation of medical devices incorporating products containing natural rubber latex February 2004 |
|
| MEDDEV 2.5/10 Guideline for Authorised Representatives January 2012 |
|
| 2.7 Clinical investigation, clinical evaluation | MEDDEV 2.7/1 rev.4 Clinical evaluation: Guide for manufacturers and notified bodies June 2016 Appendix 1: Clinical evaluation on coronary stents December 2008 |
| MEDDEV 2.7/2 rev. 2 Guidelines for Competent Authorities for making a validation/assessment of a clinical investigation application under directives 90/385/EEC and 93/42/EC September 2015 |
|
| MEDDEV 2.7/3 rev. 3 Clinical investigations: serious adverse reporting under directives 90/385/EEC and 93/42/EC - SAE reporting form May 2015 |
|
| The new SAE reporting form will be taken in use 1 September 2016 at the latest. | |
| MEDDEV 2.7/4 Guidelines on Clinical investigations: a guide for manufacturers and notified bodies December 2010 |
|
| 2.10 Notified bodies | MEDDEV 2.10/2 rev.1 Designation and monitoring of Notified Bodies within the framework of EC Directives on Medical devices Annex 1, Annex 2, Annex 3, Annex 4 April 2001 |
| 2.12 Market surveillance |
MEDDEV 2.12/1 rev.8 Ⅰ. MEDDEV 2.12/1 rev.8 – Latest Version Forms Additional Guidance Regarding the Vigilance System as outlined in MEDDEV 2.12-1 rev. 8 Active PDF forms Please note: Some browser plugins are not compatible with PDF forms. If you have problems opening these forms, please save them to your computer and open them from there. Other forms and templates EU Vigilance Pilot on Trending – Additional MIR Form Ⅱ. Device Specific Vigilance Guidance |
| MEDDEV 2.12/2 rev.2 Post Market Clinical Follow-up studies January 2012 |
|
| 2.13 Transitional period | MEDDEV 2.13 rev.1 Commission communication on the application of transitional provision of Directive 93/42/EEC relating to medical devices (OJ 98/C 242/05) August 1998 |
| As regards the transitional regime of Directive 2007/47/EC see the Interpretative Document of the Commission's services of 5 June 2009 |
|
| 2.14 IVD | MEDDEV 2.14/1 rev.2 Borderline and Classification issues. A guide for manufacturers and notified bodies January 2012 |
| MEDDEV 2.14/2 rev.1 Research Use Only products February 2004 |
|
| MEDDEV 2.14/3 rev.1 Supply of Instructions For Use (IFU) and other information for In-vitro Diagnostic (IVD)Medical Devices January 2007 |
|
| Form for the registration of manufacturers and devices In Vitro Diagnostic Medical DeviceDirective, Article 10 January 2007 |
|
| MEDDEV 2.14/4 CE marking of blood based in vitro diagnostic medical devices for vCJD based on detection of abnormal PrP January 2012 |
|
| 2.15 Other guidances | MEDDEV 2.15 rev.3 Committees/Working Groups contributing to the implementation of the Medical Device Directives December 2008 |
Matrice Revisioni Certifico:
| Rev. | Data | Oggetto | Autore |
| 34.0 | 26.02.2024 |
MDCG 2024-2 Procedures for the updates of the EMDN February 2024 |
Certifico Srl |
| 33.0 | 28.09.2023 |
- MDCG 2023-3 Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 on medical devices Febraury 2023 |
Certifico Srl |
| 32.0 | 24.01.2023 | - MDCG 2023-2 MDR form List of Standard Fees January 2023 - MDCG 2023-2 IVDR form List of Standard Fees January 2023 - MDCG 2023-2 List of Standard Fees January 2023 - MDCG 2023-1 Guidance on the health institution exemption under Article 5(5) of Regulation (EU) 2017/745 and Regulation (EU) 2017/746 January 2023 - MDCG 2022-21 Guidance on Periodic Safety Update Report (PSUR) according to Regulation (EU) 2017/745 December 2022 - MDCG 2022-20 Substantial modification of performance study under Regulation (EU) 2017/746 December 2022 - MDCG 2022-19 Performance study application/notification documents under Regulation (EU) 2017/746 December 2022 - MDCG 2022-18 MDCG Position Paper on the application of Article 97 MDR to legacy devices for which the MDD or AIMDD certificate expires before the issuance of a MDR certificate December 2022 - MDCG 2022-17 MDCG position paper on "hybrid audits" December 2022 - MDCG 2022-16 Guidance on Authorised Representatives Regulation (EU) 2017/745 and Regulation (EU) 2017/746 October 2022 - MDCG 2022-15 Guidance on appropriate surveillance regarding the transitional provisions under Article 110 of the IVDR with regard to devices covered by certificates according to the IVDD September 2022 - MDCG 2022-14 Transition to the MDR and IVDR - Notified body capacity and availability of medical devices and IVDs August 2022 - MDCG 2022-4 rev.1 Guidance on appropriate surveillance regarding the transitional provisions under Article 120 of the MDR with regard to devices covered by certificates according to the MDD or the AIMDD December 2022 |
Certifico Srl |
| 31.0 | 07.09.2022 | MDCG 2022-13 Designation, re-assessment and notification of conformity assessment bodies and notified bodies MDCG 2022-12 Guidance on harmonised administrative practices and alternative technical solutions until Eudamed is fully functional (for Regulation (EU) 2017/746 on in vitro diagnostic medical devices) |
Certifico Srl |
| 30.0 | 06.07.2022 | MDCG 2022-11 MDCG Position Paper: Notice to manufacturers to ensure timely compliance with MDR requirements MDCG 2022-10 Q&A on the interface between Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) MDCG 2022-9 Summary of safety and performance template MDCG 2022-8 Regulation (EU) 2017/746 - application of IVDR requirements to ‘legacy devices’ and to devices placed on the market prior to 26 May 2022 in accordance with Directive 98/79/EC MDCG 2022-7 Q&A on the Unique Device Identification system under Regulation (EU) 2017/745 and Regulation (EU) MDCG 2022-6 Guidance on significant changes regarding the transitional provision under Article 110(3) of the IVDR |
Certifico Srl |
| 29.0 | 20.05.2022 |
MDCG 2022 - 5 Guidance on borderline between medical devices and medicinal products under Regulation (EU) 2017/745 on medical |
Certifico Srl |
| 28.0 | 23.03.2022 |
MDCG 2022-4 Guidance on appropriate surveillance regarding the transitional provisions under Article 120 of the MDR |
Certifico Srl |
| 27.0 | 01.02.2022 |
MDCG 2022-2 Guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices (IVDs) |
Certifico Srl |
| 26.0 | 15.11.2021 |
MDCG 2021-26 - Questions and Answers on repackaging & relabelling activities under Article 16 of Regulation (EU) 2017/745 and Regulation (EU) 2017/746 |
Certifico Srl |
| 25.0 | 16.10.2021 |
MDCG 2021-24 Guidance on classification of medical devices October 2021 |
Certifico Srl |
| 24.0 | 10 Settembre 2021 |
MDCG 2021-6 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation April 2021 |
Certifico Srl |
| 23.0 | 22 Aprile 2021 |
MDCG 2021-5 Guidance on standardisation for medical devices |
Certifico Srl |
| 22.0 | 07 Luglio 2020 |
MDCG 2020-12 |
Certifico Srl |
| 21.0 | 28 Maggio 2020 |
MDCG 2020-11 |
Certifico Srl |
| 20.0 | 29 Aprile 2020 |
MDCG 2020-9 |
Certifico Srl |
| 19.0 | 26 Aprile 2020 |
MDCG 2020-5 |
Certifico Srl |
| 18.0 | 09 Aprile 2020 |
MDCG 2019-3 |
Certifico Srl |
| 17.0 | 18 Marzo 2020 |
MDCG 2020-3 |
Certifico Srl |
| 16.0 | 08 Gennaio 2020 |
Guidance on Cybersecurity for medical devices |
Certifico Srl |
| 15.0 | 18 Dicembre 2019 |
Guidance notes for manufacturers of class I medical devices |
Certifico Srl |
| 14.0 | 11 Dicembre 2019 |
- MDCG 2019-14 Explanatory note on MDR codes |
Certifico Srl |
| 13.0 | 30 Novembre 2019 |
Qualification and classification of software |
Certifico Srl |
| 12.0 | 08 Ottobre 2019 | MDCG 2019-10 Application of transitional provisions concerningvalidity of certificates issued in accordance to Directives 90/ 385/EEC and 93/42/EEC |
Certifico Srl |
| 11.0 | 27 Settembre 2019 | MDCG 2019-9 Summary of safety and clinical performance A guide for manufacturers and notified bodies DSVG 03 - Cardiac Implantable Electronic Devices (CIED) Guidance vigilance system for CE-marked medical devices DSVG 04 - Breast Implants Guidance vigilance system for CE-marked medical devices |
Certifico Srl |
| 10.0 | 12 Luglio | MDCG Documents Additional Guidance Regarding the Vigilance System MEDDEV 2.12-1 rev. 8 |
Certifico Srl |
| 9.0 | 16 Aprile 2019 | MDCG Documents | Certifico Srl |
| 8.0 | 22 Marzo 2019 | MDCG Documents | Certifico Srl |
| 7.0 | 19 Febbraio 2019 | MDCG Documents | Certifico Srl |
| 6.0 | Febbraio 2019 | MDCG Documents | Certifico Srl |
| 5.0 | Gennaio 2019 | MDCG Documents | Certifico Srl |
| 4.0 | Ottobre 2018 | MDCG Documents | Certifico Srl |
| 3.0 | Agosto 2018 | MDCG Documents | Certifico Srl |
| 2.0 | Agosto 2016 | MEDDEV 2.1/6 MEDDEV 2.7/1 rev.4 DSVG Template DSVG 00 DSVG 01 DSVG 02 |
Certifico Srl |
| 1.0 | Novembre 2015 | ---- | Certifico Srl |
Collegati
[box-note]Regolamento (UE) 2017/745
Direttiva Dispositivi medici 93/42/CEE[/box-note]

Report 34 del 26/08/2016 N.2 A12/1071/16 Ungheria
Approfondimento tecnico: Seggiolino da bicicletta per bambini

Il prodotto, di marca Kross/bg, Mod. BG-5, è stato sottoposto alla procedura di ritiro dal mercato perché non conforme alla norma europea EN 14344.
Il sistema di bloccaggio risulta essere inadeguato. A seguito di un incidente, pertanto, il seggiolino potrebbe staccarsi dalla bicicletta.
La norma EN 14344 stabilisce, al capitolo 8, che, per evitare il rilascio accidentale del seggiolino, il sistema di bloccaggio deve richiedere:
a) l'uso di un utensile (chiave inglese o cacciavite ad esempio) da utilizzare su almeno uno dei dispositivi presenti;
b) la presenza di due meccanismi di bloccaggio che vengano azionati contemporaneamente;
c) due o più dispositivi di bloccaggio automatici che non possano essere rilasciati contemporaneamente;
d) due azioni successive per lo sblocco, la prima della quali deve essere mantenuta mentre si agisce sulla seconda.
Report 33 del 19/08/2016 N.6 A12/1020/16 Lituania
Approfondimento tecnico: Orologio da corsa

Il prodotto Forerunner 15, di marca “Garmin”, è stato richiamato dal mercato direttamente dal Fabbricante.
Le saldature di alcuni componenti interni all’orologio contengono una quantità eccessiva di piombo. Inoltre, tali componenti, sigillati all’interno del prodotto, non possono essere rimossi fintanto che il prodotto non venga smontato.
Il prodotto non è conforme alla Direttiva 2011/65/UE del Parlamento Europeo e del Consiglio dell’8 giugno 2011 sulla restrizione dell’uso di determinate sostanze pericolose nelle apparecchiature elettriche ed elettroniche (RoHS 2).
La Direttiva 2011/65/UE stabilisce che:
Articolo 4
Prevenzione
1. Gli Stati membri provvedono affinché le AEE immesse sul mercato, compresi i cavi e i pezzi di ricambio destinati alla loro riparazione, al loro riutilizzo, all’aggiornamento delle loro funzionalità o al potenziamento della loro capacità, non contengano le sostanze di cui all’allegato II.
2. Ai fini della presente direttiva nei materiali omogenei è tollerata una concentrazione massima in peso non superiore a quella indicata nell’allegato II. La Commissione adotta, mediante atti delegati conformemente all’articolo 20 e fatte salve le condizioni di cui agli articoli 21 e 22, le modalità dettagliate per garantire la conformità ai predetti valori massimi di concentrazione, anche tenendo conto dei rivestimenti superficiali. […]
ALLEGATO II
Sostanze con restrizioni d’uso di cui all’articolo 4, paragrafo 1, e valori delle concentrazioni massime tollerate per peso nei materiali omogenei
Piombo (0,1 %)
Mercurio (0,1 %)
Cadmio (0,01 %)
Cromo esavalente (0,1 %)
Bifenili polibromurati (PBB) (0,1 %)
Eteri di difenile polibromurato (PBDE) (0,1 %)

Rev. 1.0 Giugno 2017
Il Focus, aggiornamento del precedente 2013 (di cui sotto), analizza, con FAQ e Schemi gli obblighi del Regolamento sui Prodotti da Costruzione CPR 305/2011 a 4 anni dall'entrata in vigore 1° Luglio 2013.
Il CPR Regolamento Prodotti da Costruzione (UE) N. 305/2011, ha sostituito la Direttiva 89/106/CEE sui prodotti da costruzione (CPD) e fissa condizioni armonizzate per la commercializzazione dei prodotti da costruzione.
Il CPR fornisce ulteriori chiarimenti sui concetti e l’applicazione della marcatura CE; introduce procedure semplificate, che ridurranno i costi sostenuti dalle imprese, in particolare delle piccole e medie imprese (PMI). La dichiarazione di prestazione (DoP) è il concetto chiave del regolamento sui prodotti da costruzione (CPR).
La DoP dà al fabbricante la possibilità di fornire le informazioni relative sulle caratteristiche essenziali del prodotto che vuole offrire al mercato. Il fabbricante redige una dichiarazione di prestazione quando un prodotto è coperto da una norma armonizzata (EN) o una Valutazione Tecnica Europea (ETA) rilasciata da un Organismo di Valutazione Tecnica (TAB) e, di conseguenza, può essere immettere il prodotto sul mercato.
Il fabbricante, rilasciando la DoP, si assume la responsabilità della conformità del prodotto da costruzione alla dichiarazione di prestazione.
Sulla base delle informazioni contenute nel DoP, gli utenti potranno decidere l’acquisto del prodotto in base all'uso riportato e si assumeranno la piena responsabilità di tale decisione.
La DoP costituisce quindi l'elemento chiave per il funzionamento del mercato interno dei prodotti da costruzione, fornendo la necessaria trasparenza e stabilendo un chiaro sistema di ripartizione delle responsabilità tra i soggetti.
Acronimi
(TAB) Technical Assessment Body
Organismi di valutazione tecnica, facenti parte dell’Organizzazione dei TAB (TABs).
(ETA) European Technical Assessment
Valutazione Tecnica Europea.
(EAD) European Assessment Document
Documento per la Valutazione Europea che è adottato dall'Organizzazione dei TABs (Technical Assessment Bodies) ai fini del rilascio delle Valutazioni Tecniche Europee ETAs (European Technical Assessments).
(ATD) Appropriate Technical Documentation
Documentazione che il fabbricante ritiene opportuno produrre, al fine di giustificare le modalità per dichiarare la prestazione del prodotto nei casi previsti dall'articolo 36 del CPR.
(DoP) Declaration of Performance
Dichiarazione di Prestazione
(AVCP) Assessment and Verification of Constancy of Performance
Le norme tecniche armonizzate o i documenti europei di valutazione (EAD), contengono le attività specifiche (compiti) necessarie per gli organismi notificati, al fine di garantire la valutazione e verifica della costanza della prestazione
Indice
1. Prodotto da costruzione da immettere sul mercato dopo il 30/06/2013
2. Prodotto da costruzione gia’ immesso sul mercato prima del 01/07/2013
3. Obblighi dei distributori
4. Immissione sul mercato
5. Obbligo rinnovo rapporti di prova, valutazioni, ecc.
6. Benestare Tecnico Europeo (ETA - European Technical Approval)
7. Prodotto da costruzione non coperto da norma armonizzata e costruito dopo il 01.07.2013
8. Documentazione tecnica appropriata
9. Clausole di norma armonizzata contrastanti con il cpr
10. Contact point for construction
11. Gli Organismi Notificati: sorveglianza e controllo
12. Dichiarazione di Prestazione (DoP) in formato elettronico
13. Regole e condizioni per l'apposizione della marcatura ce
14. Manuale di installazione/istruzioni
I. Blibliografia
Abbonati: Marcatura CE/2X/3X/4X/Full
Certifico Srl IT - Rev. 00 2017
Maggiori Info e acquisto Documento
Documento compreso nel Servizio di Abbonamento Marcatura CE
__________
Precedenti Ed. 2013

CPR UE 305/2011 Dichiarazione di Prestazione - DoP
Regolamento Prodotti da Costruzione (UE) 305/2011 - CPR
Direttiva Prodotti da costruzione 89/106/CEE
_____________
SINTESI
Il presente regolamento determina le condizioni relative all'immissione sul mercato dei prodotti da costruzione. Definisce anche criteri di valutazione delle prestazioni per questi prodotti e le condizioni di utilizzo della marcatura CE.
Dichiarazione di Prestazione e marcatura CE
Qualora il fabbricante decida di immettere sul mercato un prodotto da costruzione che rientra nell'ambito di applicazione di una norma armonizzata, deve compilare una dichiarazione di prestazione dove saranno riportate soprattutto le informazioni seguenti:
- il riferimento del prodotto;
- i sistemi di valutazione e verifica della costanza della prestazione del prodotto;
- l'uso o gli usi previsti del prodotto;
- la prestazione dichiarata.
Una volta redatta la dichiarazione di prestazione, il fabbricante deve apporre la marcatura CE sul prodotto.
Gli Stati membri designano punti di contatto di prodotti da costruzione in conformità con il regolamento (CE) n. 764/2008. Questi punti di contatto devono fornire informazioni relative ai requisiti del prodotto da costruzione ed evitare i conflitti d'interesse.
Obblighi degli operatori economici
Agli operatori economici vengono imposti determinati obblighi:
Obblighi dei fabbricanti: devono fornire la dichiarazione di prestazione e la documentazione tecnica, ed apporre la marcatura CE sul prodotto. I fabbricanti assicurano che i loro prodotti rechino un numero di tipo che consenta la loro identificazione. Essi sono inoltre tenuti a ritirare i loro prodotti dal mercato, se ritengono che non siano conformi alla dichiarazione di prestazione, o a cambiare questa dichiarazione.
Obblighi degli importatori: verificano che il prodotto sia accompagnato dalla documentazione tecnica e che rechi la marcatura CE. Essi devono indicare il loro nome, la loro denominazione commerciale registrata o il loro marchio registrato e l'indirizzo cui possono essere contattati. Essi assicurano che il prodotto sia accompagnato da istruzioni ed informazioni sulla sicurezza e che il trasporto non alteri la sua prestazione.
Obblighi dei distributori: devono assicurarsi che il prodotto rechi la marcatura CE e che sia accompagnato dai documenti di cui sopra. Qualora ritengano che il prodotto non è conforme, devono astenersi dall'immetterlo sul mercato. I distributori devono inoltre garantire condizioni ottimali di conservazione del prodotto affinché non si degradi.
Specifiche tecniche armonizzate
Le specifiche tecniche armonizzate comprendono le norme armonizzate. Esse sono stabilite dalle organizzazioni europee di normalizzazione conformemente alla direttiva 98/34/CE. Le norme armonizzate servono a definire i metodi ed i criteri di valutazione delle prestazioni dei prodotti da costruzione. Esse si riferiscono all'uso previsto dei prodotti cui si riferiscono e includono i dettagli tecnici necessari per applicare il sistema di valutazione e verifica della costanza della prestazione. I riferimenti alle norme armonizzate sono pubblicati nella Gazzetta ufficiale dell’Unione europea.
Se un prodotto non è coperto da una norma armonizzata, un fabbricante ha la possibilità di chiedere una valutazione tecnica europea per ottenere un documento di valutazione europeo redatto dall’organizzazione degli organismi di valutazione tecnica (TAB).
Organismi di valutazione tecnica (TAB)
Gli Stati membri designano uno o più TAB sul loro territorio, per una o più aree di prodotto. L'elenco dei TAB viene comunicato alla Commissione europea che lo pubblica.
I TAB effettuano la valutazione tecnica europea in un'area di prodotto per la quale sono stati designati.
Autorità notificanti e organismi notificati
Gli organismi notificati svolgono compiti di parte terza nel processo di valutazione e verifica della costanza della prestazione dei prodotti da costruzione. Si tratta di organismi indipendenti dotati di personalità giuridica.
Le autorità notificanti sono poste in essere dagli Stati membri. Esse sono responsabili della creazione ed applicazione delle procedure necessarie per la valutazione e la notifica degli organismi notificati.
Vigilanza del mercato
Le autorità di vigilanza del mercato, conformemente anche al regolamento (CE) n. 765/2008, devono effettuare una valutazione del prodotto per determinare se è opportuno o meno ritirarlo dal mercato.
Il presente regolamento abroga la direttiva 89/106/CEE.
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| Il CPR Regolamento Prodotti da Costruzione UE 305 2011 Rev. 1.0 2017.pdf Certifico Srl - Rev. 1.0 2017 |
1199 kB | 269 | ||
| Il CPR Regolamento Prodotti da Costruzione UE 305 2011 Rev. 0.0 2013.pdf Certifico Srl - Rev. 0.0 2013 |
1395 kB | 92 |
Report 31 del 05/08/2016 N.1 A12/0929/16 Italia
Approfondimento tecnico: Liquido per sigarette elettroniche

Il prodotto, di marca “Quickie”, è stato sottoposto al divieto di immissione sul mercato, commercializzazione, importazione, fabbricazione ed esportazione ai sensi dell’art. 5 del D.Lgs. 25 gennaio 1992, n. 73, ed al divieto di commercializzazione del Ministero della Salute prot. 0012486-06/05/2016-DGPRE-COD_UO-P, a seguito di esito della valutazione di un campione il 4 maggio 2016 presso il Ministero della Salute con la presenza di un rappresentante dell’ISS.
La ricarica per sigaretta elettronica veniva venduta in un contenitore in plastica di forma cilindrica con base ellittica di colore giallo, recante in etichetta adesiva l’indicazione “QUICKIE- Milk and Chocolate 100% Natural flavors 6 mg” e sulla base alta piccola etichetta adesiva recante l’indicazione “contiene Nicotina. Nocivo a contatto con la pelle. Tenere fuori dalla portata dei bambini”, con all’interno n.1 (uno) flacone contenente ricarica per sigaretta elettronica e n.2 (due) flaconi vuoti.
Il liquido non è risultato conforme al Regolamento (UE) 1272/2008 (CLP) ed alla Direttiva 87/357/CEE.
Il Regolamento (UE) 1272/2008 (CLP) stabilisce che:
Articolo 33
Disposizioni particolari relative all'etichettatura dell'imballaggio esterno, dell'imballaggio interno e dell'imballaggio unico
1. Quando un collo comprende un imballaggio esterno e uno interno nonché un eventuale imballaggio intermedio e l'imballaggio esterno è conforme alle disposizioni in materia di etichettatura previste dalle norme per il trasporto di merci pericolose, l'imballaggio interno e l'eventuale imballaggio intermedio sono etichettati in conformità del presente regolamento. Anche l'imballaggio esterno può essere etichettato conformemente al presente regolamento. Nei casi in cui il pittogramma o i pittogrammi di pericolo previsti dal presente regolamento si riferiscono allo stesso pericolo contemplato dalle norme per il trasporto di merci pericolose, il pittogramma o i pittogrammi di pericolo previsti dal presente regolamento possono non figurare sull'imballaggio esterno.
2. Quando l'imballaggio esterno di un collo non è soggetto alle disposizioni in materia di etichettatura previste dalle norme per il trasporto di merci pericolose, sia l'imballaggio esterno che quello interno, nonché l'eventuale imballaggio intermedio, sono etichettati conformemente al presente regolamento. Tuttavia, se l'imballaggio esterno permette di vedere chiaramente l'etichettatura dell'imballaggio interno o di quello intermedio, l'imballaggio esterno può non essere etichettato.
3. I colli unici conformi alle disposizioni in materia di etichettatura previste dalle norme per il trasporto di merci pericolose sono etichettati conformemente alle disposizioni del presente regolamento e alle norme in materia di trasporto delle merci pericolose. Nei casi in cui il pittogramma o i pittogrammi di pericolo previsti dal presente regolamento si riferiscono allo stesso pericolo contemplato dalle norme in materia di trasporto di merci pericolose, il pittogramma o i pittogrammi di pericolo previsti dal presente regolamento possono non figurare.
Inoltre, non risultando espresso in percentuale il contenuto di nicotina, non è possibile verificare la corretta classificazione di pericolo e la conseguente etichettatura.
Per quanto riguarda la Direttiva 87/357/CEE, invece:
Articolo 1
1. La presente direttiva si applica ai prodotti definiti nel paragrafo 2 che, avendo un aspetto diverso da quello che sono in realtà, compromettono la sicurezza o la salute dei consumatori.
2. I prodotti di cui al paragrafo 1 sono quelli che, pur non essendo prodotti alimentari, hanno forma, odore, aspetto, imballaggio, etichettatura, volume o dimensioni tali da far prevedere che i consumatori, soprattutto i bambini, li possono confondere con prodotti alimentari e pertanto li portino alla bocca, li succhino o li ingeriscano con conseguente rischio di soffocamento, intossicazione, perforazione o ostruzione del tubo digerente.
Articolo 2
Gli Stati membri adottano tutte le misure necessarie perché sia vietata la commercializzazione, l'importazione, la fabbricazione e l'esportazione dei prodotti di cui alla presente direttiva.
Infine, con il D.Lgs. 12 gennaio 2016 n. 6, recepimento della Direttiva 2014/40/UE, dal 20 maggio 2016 non è più consentito vendere in Italia liquidi di ricarica in contenitori di volume superiore a 10 ml. Il contenitore in oggetto conteneva invece 100 ml di prodotto.

QUADRI ELETTRICI PER BORDO MACCHINA Caratteristiche, prescrizioni e normative
Guida Tecnica
La presente guida è dedicata ai costruttori e agli assemblatori di Quadri bordo macchina.
Le norme tecniche e le direttive in vigore applicabili a questo comparto sono molteplici e rappresentano le linee guida alle quali attenersi per la progettazione e la realizzazione di apparecchiature con elevatissimi standard di sicurezza e affidabilità sia in fase di utilizzo che in fase di manutenzione preventiva.
L’obiettivo della guida è quello di spiegare in maniera dettagliata e attraverso esempi pratici:
- i contenuti delle norme e delle direttive;
- le caratteristiche tecniche dei quadri bordo macchina;
- i materiali utilizzati per la loro costruzione e utilizzo nei diversi ambienti produttivi;
- i ruoli e le responsabilità dei costruttori e assemblatori.
La guida è stata realizzata dal Gruppo di Lavoro “Quadri Bordo Macchina” di ANIE Energia.
Le principali attività del gruppo riguardano il monitoraggio del mercato, la normazione tecnica attraverso una costante partecipazione attiva a comitati e organi tecnici e progetti di comunicazione.
ANIE 2016
Certifico Equipaggiamenti Elettrici EN 60204-1 Ed. 2016
Direttiva 2010/30/UE del Parlamento europeo e del Consiglio del 19 maggio 2010 concernente l’indicazione del consumo di energia e di altre risorse dei prodotti connessi all’energia, mediante l’etichettatura ed informazioni uniformi relative ai prodotti.
GU L 153/1 del 18.6.2010
Abrogazione: la direttiva 2010/30/UE è abrogata a decorrere dal 1° agosto 2017 dal Regolamento (UE) 2017/1369
Collegati:
[box-note]Regolamento (UE) 2017/1369
Ecodesign Energy Labelling
[/box-note]

ID 2843 | 18.02.2025 / In allegato
Decreto Legislativo 4 luglio 2014 n. 102
Attuazione della direttiva 2012/27/UE sull’efficienza energetica, che modifica le direttive 2009/125/CE e 2010/30/UE e abroga le direttive 2004/8/CE e 2006/32/CE.
(GU n. 165 del 18 Luglio 2014)
Allegati:
- Testo nativo
- Testo consolidato 2018
- Testo consolidato 2020
- Testo consolidato 2022
- Testo consolidato 2023
- Testo consolidato 2024
- Testo consolidato 2025
_________
[box-note]Aggiornamenti all'atto
- Avviso di rettifica (in G.U. 24/07/2014, n.170)
- Decreto-Legge 12 settembre 2014, n. 133 (in G.U. 12/09/2014, n.212), convertito con modificazioni dalla L. 11 novembre 2014, n. 164 (in S.O. n. 85, relativo alla G.U. 11/11/2014, n. 262) - Ed 1.0 Marzo 2018
- Decreto-Legge 30 dicembre 2015, n. 210 (in G.U. 30/12/2015, n.302) , convertito con modificazioni dalla L. 25 febbraio 2016, n. 21 (in G.U. 26/02/2016, n. 47) - Ed 1.0 Marzo 2018
- Decreto Legislativo 18 luglio 2016, n. 141 (in G.U. 25/07/2016, n.172) - Ed 1.0 Marzo 2018
- Decreto-Legge 29 dicembre 2016, n. 243 (in G.U. 30/12/2016, n.304) convertito con modificazioni dalla L. 27 febbraio 2017, n. 18 (in G.U. 28/02/2017, n.49) - Ed 1.0 Marzo 2018
- Legge 27 dicembre 2017, n. 205 (in SO n.62, relativo alla G.U. 29/12/2017, n.302) - Ed 1.0 Marzo 2018
- Decreto Legislativo 10 giugno 2020 n. 48 (in GU Serie Generale n.146 del 10-06-2020) - Ed. 2.0 Giugno 2020
- Decreto Legislativo 14 luglio 2020 n. 73 (in GU Serie Generale n.175 del 14-07-2020) - Ed. 3.0 Luglio 2020
- Legge 30 dicembre 2021 n. 234 (in SO n.49, relativo alla G.U. 31/12/2021 n.310) - Ed. 4.0 Febbraio 2022
- Decreto-Legge 1 marzo 2022 n. 17 (GU n.50 del 01.03.2022) - Ed. 5.0 aprile 2022
- Legge 27 aprile 2022 n. 34 (GU n.98 del 28.04.2022) - Ed. 5.1 aprile 2022
- Decreto-Legge 24 febbraio 2023, n. 13 (G.U. n. 47 del 24.02.2023) - Ed. 6.0 ottobre 2023
- Legge 30 dicembre 2023 n. 214 (GU n.303 del 30.12.2023) - Ed. 7.0 Gennaio 2024
- Legge 16 dicembre 2024 n. 193 (GU n.295 del 17.12.024) - Ed. 8.0 Gennaio 2025[/box-note]
_________
Il presente decreto, in attuazione della direttiva 2012/27/UE e nel rispetto dei criteri fissati dalla legge 6 agosto 2013, n. 96, stabilisce un quadro di misure per la promozione e il miglioramento dell’efficienza energetica che concorrono al conseguimento dell’obiettivo nazionale di risparmio energetico indicato all’articolo 3 (Obiettivo nazionale di risparmio energetico).
Il presente decreto, inoltre, detta norme finalizzate a rimuovere gli ostacoli sul mercato dell’energia e a superare le carenze del mercato che frenano l’efficienza nella fornitura e negli usi finali dell’energia.
...
Art. 8. Diagnosi energetiche e sistemi di gestione dell’energia
1. Le grandi imprese eseguono una diagnosi energetica, condotta da società di servizi energetici, esperti in gestione dell’energia o auditor energetici e da ISPRA relativamente allo schema volontario EMAS, nei siti produttivi localizzati sul territorio nazionale entro il 5 dicembre 2015 e successivamente ogni 4 anni, in confor- mità ai dettati di cui all’allegato 2 al presente decreto. Tale obbligo non si applica alle grandi imprese che hanno adottato sistemi di gestione conformi EMAS e alle norme ISO 50001 o EN ISO 14001, a condizione che il sistema di gestione in questione includa un audit energetico realizzato in conformità ai dettati di cui all’allegato 2 al presente decreto. I risultati di tali diagnosi sono comunicati all’ENEA e all’ISPRA che ne cura la conservazione.
2. Decorsi 24 mesi dalla data di entrata in vigore del presente decreto, le diagnosi di cui al comma 1 sono eseguite da soggetti certificati da organismi accreditati ai sensi del regolamento comunitario n. 765 del 2008 o firmatari degli accordi internazionali di mutuo riconoscimento, in base alle norme UNI CEI 11352, UNI CEI 11339 o alle ulteriori norme di cui all’articolo 12, comma 3, relative agli auditor energetici, con l’esclusione degli installatori di elementi edilizi connessi al miglioramento delle prestazioni energetiche degli edifici. Per lo schema volontario EMAS l’organismo preposto è ISPRA.
3. Le imprese a forte consumo di energia che ricadono nel campo di applicazione dell’articolo 39, comma 1 o comma 3, del decreto-legge 22 giugno 2012, n. 83, convertito, con modificazioni, dalla legge 7 agosto 2012, n. 134, sono tenute, ad eseguire le diagnosi di cui al comma 1, con le medesime scadenze, indipendentemente dalla loro dimensione e a dare progressiva attuazione, in tempi ragionevoli, agli interventi di efficienza individuati dalle diagnosi stesse o in alternativa ad adottare sistemi di gestione conformi alle norme ISO 50001.
4. Laddove l’impresa soggetta a diagnosi sia situata in prossimità di reti di teleriscaldamento o in prossimità di impianti cogenerativi ad alto rendimento, la diagnosi contiene anche una valutazione della fattibilità tecnica, della convenienza economica e del beneficio ambientale, derivante dall’utilizzo del calore cogenerato o dal collegamento alla rete locale di teleriscaldamento.
5. L’ENEA istituisce e gestisce una banca dati delle imprese soggette a diagnosi energetica nel quale sono riportate almeno l’anagrafica del soggetto obbligato e dell’auditor, la data di esecuzione della diagnosi e il rapporto di diagnosi.
6. L’ENEA svolge i controlli che dovranno accertare la conformità delle diagnosi alle prescrizioni del presente articolo, tramite una selezione annuale di una percentuale statisticamente significativa della popolazione delle imprese soggetta all’obbligo di cui ai commi 1 e 3, almeno pari al 3%. ENEA svolge il controllo sul 100 per cento delle diagnosi svolte da auditor interni all’impresa. L’attività di controllo potrà prevedere anche verifiche in situ.
7. In caso di inottemperanza riscontrata nei confronti dei soggetti obbligati, si applica la sanzione amministrativa di cui al comma 1 dell’articolo 16.
8. Entro il 30 giugno di ogni anno ENEA, a partire dall’anno 2016, comunica al Ministero dello sviluppo economico e al Ministero dell’ambiente, della tutela del territorio e del mare, lo stato di attuazione dell’obbligo di cui ai commi 1 e 3 e pubblica un rapporto di sintesi sulle attività diagnostiche complessivamente svolte e sui risultati raggiunti.
9. Entro il 31 dicembre 2014 il Ministero dello sviluppo economico, di concerto con il Ministero dell’ambiente, della tutela del territorio e del mare, pubblica un bando per il cofinanziamento di programmi presentati dalle Regioni finalizzati a sostenere la realizzazione di diagnosi energetiche nelle PMI o l’adozione nelle PMI di sistemi di gestione conformi alle norme ISO 50001. I programmi di sostegno presentati dalle Regioni prevedono che gli incentivi siano concessi alle imprese beneficiarie nel rispetto della normativa sugli aiuti di Stato e a seguito della effettiva realizzazione delle misure di efficientamento energetico identificate dalla diagnosi energetica o dell’ottenimento della certificazione ISO 50001.
10. All’attuazione delle attività previste al comma 9 si provvede, nel limite massimo di 15 milioni di euro per ciascuno degli anni dal 2014 al 2020, a valere sulla quota spettante al Ministero dello sviluppo economico dei proventi annui delle aste delle quote di emissione di CO2 di cui all’articolo 19 del decreto legislativo 13 marzo 2013, n. 30, destinati ai progetti energetico ambientali, con le modalità e nei limiti di cui ai commi 3 e 6 dello stesso articolo 19, previa verifica dell’entità dei proventi disponibili annualmente.
11. All’attuazione delle attività previste ai commi 5 e 6 del presente articolo si provvede nel limite massimo di 0,3 milioni di euro per ciascuno degli anni dal 2014 al 2020, a valere sulla quota spettante al Ministero dello sviluppo economico dei proventi annui delle aste delle quote di emissione di CO2 di cui all’articolo 19 del decre- to legislativo 13 marzo 2013, n. 30, destinati ai progetti energetico ambientali, con le modalità e nei limiti di cui ai commi 3 e 6 dello stesso articolo 19, previa verifica dell’entità dei proventi disponibili annualmente.
Entrata in vigore del provvedimento: 19/07/2014
Vedi il Testo consolidato:

Collegati
[box-note]Decreto Legislativo 10 giugno 2020 n. 48
Decreto Legislativo 14 luglio 2020 n. 73
D.Lgs. 192/2005 Efficienza energetica edilizia | Consolidato
Grande impresa: definizione e obblighi energetici D. Lgs. 102/2004
Direttiva 2009/125/CE
Direttiva 2010/30/UE
Direttiva 2012/27/UE
Decreto 16 settembre 2016
Decreto 17 luglio 2014 | PAEE 2014
Decreto 11 dicembre 2017 | PAEE 2017[/box-note]

Direttiva 2006/42/CE: Classificazione attrezzature utilizzate per il sollevamento di carichi con macchine per il sollevamento
Questa classificazione è stata approvata dal "Gruppo di lavoro macchine" come base per una coerente applicazione del termine "accessorio di sollevamento" come definito nella Articolo 2(d)*, della Direttiva Macchine 2006/42/CE.
Il documento fornisce esempi di attrezzature che sono considerate accessori di sollevamento e altri esempi di attrezzature utilizzate per il sollevamento di carichi che non sono considerate accessori di sollevamento.
Aggiornamento: Giugno 2012
Commissione europea
(*)Direttiva macchine 2006/42/CE Articolo 2 d) "accessori di sollevamento":
componenti o attrezzature non collegate alle macchine per il sollevamento, che consentono la presa del carico, disposti tra la macchina e il carico oppure sul carico stesso, oppure destinati a divenire parte integrante del carico e ad essere immessi sul mercato separatamente. Anche le imbracature e le loro componenti sono considerate accessori di sollevamento.
Directive 2006/42/EC Classification of equipment used for lifting loads with lifting machinery
This classification was approved by the Machinery Working Group as a basis for a consistent application of the term ‘lifting accessory’ as defined in
Article 2(d)* of the Machinery Directive 2006/42/EC.
The document gives examples of equipment that are considered as lifting accessories and other examples of equipment used for lifting loads that are not considered as lifting accessories.
Update: June 2012
European Commission
(*)Machinery Directive 2006/42/EC Article 2(d) ‘"lifting accessory"
means a component or equipment not attached to the lifting machinery, allowing the load to be held, which is placed between the machinery and the load or on the load itself, or which is intended to constitute an integral part of the load and which is independently placed on the market; slings and their components are also regarded as lifting accessories.
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| Classication_equipment_lifting_loads_Directive_2006_42_EC.pdf Directive 2006/42/EC |
1506 kB | 873 |
Report 29 del 22/07/2016 N.5 A12/0898/16 Spagna
Approfondimento tecnico: Frigorifero

Il prodotto “Frostfri”, di marca IKEA, è stato ritirato dal mercato, dietro iniziativa del Fabbricante, perché non conforme alla Direttiva 2006/95/CE Bassa Tensione (ad oggi Direttiva 2014/35/UE).
Il prodotto presenta un rischio di scossa elettrica a causa del pannello orizzontale, posto dietro lo sportello nella parte superiore dell’elettrodomestico, che può allentarsi o staccarsi esponendo le persone al rischio di contatto con parti sotto tensione elettrica.
Ikea ha comunicato ai propri clienti, in possesso di un frigorifero e/o congelatore Frostfri prodotti tra la settimana 45 del 2015 e la settimana 7 del 2016, di non utilizzarli (staccando la spina di alimentazione) e di fissare un appuntamento per la riparazione.
Per capire in quale settimana è stato prodotto il proprio frigorifero e/o congelatore basta leggere la data riportata sull’etichetta accessibile rimuovendo il cassetto inferiore. La data è rappresentata da 4 numeri nell’angolo in alto a destra dell’etichetta.
I prodotti sono stati venduti in Svezia, Norvegia, Danimarca, Finlandia, Islanda, Germania, Spagna, Francia e Italia.
Per essere conforme alla Direttiva 2006/95/CE, un prodotto deve rispettare quanto riportato nell’Allegato I dello stessa:
ALLEGATO I
Elementi Principali degli Obiettivi di Sicurezza del Materiale Elettrico Destinato ad essere Adoperato entro taluni Limiti di Tensione
1. Requisiti generali
a) Le caratteristiche essenziali del materiale elettrico, la cui conoscenza ed osservanza sono indispensabili per un impiego conforme alla destinazione ed esente da pericolo, sono indicate sul materiale elettrico stesso oppure, qualora ciò non sia possibile, su una scheda che l'accompagna.
b) Il marchio di fabbrica o il marchio commerciale sono apposti distintamente sul materiale elettrico oppure, se ciò non è possibile, sull'imballaggio.
c) Il materiale elettrico e le sue parti costitutive sono costruiti in modo da poter essere collegati in maniera sicura ed adeguata.
d) Il materiale elettrico è progettato e fabbricato in modo da assicurare la protezione dai pericoli citati ai punti 2 e 3 del presente allegato, sempreché esso sia adoperato in conformità della sua destinazione e osservando le norme di manutenzione.
2. Protezione dai pericoli che possono derivare dal materiale elettrico
In conformità al punto 1 sono previste misure di carattere tecnico affinché:
a) le persone e gli animali domestici siano adeguatamente protetti dal pericolo di ferite o altri danni che possono derivare da contatti diretti o indiretti;
b) non possano prodursi sovratemperature, archi elettrici o radiazioni che possano causare un pericolo;
c) le persone, gli animali domestici e gli oggetti siano adeguatamente protetti dai pericoli di natura non elettrica che, come insegna l'esperienza, possono derivare dal materiale elettrico;d) l'isolamento sia proporzionato alle sollecitazioni previste.
3. Protezione dai pericoli dovuti all'influenza di fattori esterni sul materiale elettrico
In conformità del punto 1, sono previste misure di ordine tecnico affinché il materiale elettrico:
a) presenti le caratteristiche meccaniche richieste in modo da non causare pericolo alle persone, agli animali domestici e agli oggetti;
b) sia resistente a fenomeni di natura non meccanica nelle condizioni ambientali previste, in modo da non causare pericolo alle persone, agli animali domestici e agli oggetti;
c) nelle condizioni di sovraccarico previste, non causi pericolo alle persone, agli animali domestici e agli oggetti.
L’unica differenza tra la Direttiva 2006/95/CE e la nuova Direttiva 2014/35/UE, in riferimento all’Allegato I, è la rimozione, nella nuova versione, del punto:
b) Il marchio di fabbrica o il marchio commerciale sono apposti distintamente sul materiale elettrico oppure, se ciò non è possibile, sull'imballaggio.

Documento Tecnico
Lista di riscontro dei metodi di verifica e validazione dei requisiti di sicurezza
Estratto EN ISO 10218-1:2011
Robot e attrezzature per robot - Requisiti di sicurezza per robot industriali - Parte 1: Robot
La norma specifica i requisiti e le linee guida per la sicurezza integrata nella progettazione, le misure protettive e l’informazione per l’uso dei robot industriali. Essa descrive i pericoli di base associati a tali robot e fornisce i requisiti per eliminare, o ridurre adeguatamente, i rischi associati a tali pericoli.
I metodi di Verifica e Validazione dei Requisiti di Sicurezza previsti dalla norma sono:
A - ispezione visiva
B - prove pratiche
C - misure
D - osservazione durante le operazioni
E - revisione di specifiche schematiche, diagrammi circuitali e materiali utilizzati
F - revisione della valutazione del rischio
G - revisione di specifiche e informazioni per l'uso
Tali metodi sono tabellati per i Requisiti di sicurezza previsti dalla norma in Appendice normativa F.
Elaborazione Certifico Rev. 01 2012
Pag. 26 - comprensivo di estratto p. 5,6,7 norma armonizzata EN ISO 10218-1:2011
Maggiori info e acquisto Documento singolo
Documento compreso nel Servizio di Abbonamento Marcatura CE
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| DT EN ISO 10218-1 Robot Check list Verifiche e Validazioni.pdf Documento Tecnico EN ISO 10218-1 |
319 kB | 370 |
Report 28 del 15/07/2016 N.23 A12/0857/16 Germania
Approfondimento tecnico: Automobile giocattolo

Il prodotto, di marca Kids Fun, Mod. 8372, è stato ritirato dal mercato perché non conforme al Regolamento n. 1907/2006 del Parlamento Europeo e del Consiglio del 18 dicembre 2006 concernente la registrazione, la valutazione, l'autorizzazione e la restrizione delle sostanze chimiche (REACH).
I pneumatici della vettura contengono idrocarburi policiclici aromatici (IPA) in concentrazioni che vanno da 9 a 179 mg/kg. Gli idrocarburi policiclici aromatici (IPA) sono cancerognei ed il bambino può esserne esposto nel momento in cui i pneumatici vengono a contatto diretto e prolungato con la pelle o la bocca.
L’Allegato XVII del Regolamento n. 1907/2006 (REACH), concernente le restrizioni in materia di fabbricazione, immissione sul mercato e uso di talune sostanze, miscele e articoli pericolosi, stabilisce, per gli idrocarburi policiclici aromatici (IPA), che:
1. A decorrere dal 1 gennaio 2010, non possono essere immessi sul mercato o utilizzati per la produzione di pneumatici o parti di pneumatici gli olii diluenti aventi un contenuto:
- di BaP superiore a 1 mg/kg (0,0001 % in peso), o
- un contenuto complessivo di tutti gli IPA elencati superiore a 10 mg/kg (0,001 % in peso).
La norma EN 16143-2013 [Prodotti petroliferi — Determinazione del contenuto di Benzo(a)pirene (BaP) e di alcuni idrocarburi policiclici aromatici (IPA) negli oli diluenti — Procedimento che utilizza la doppia purificazione mediante LC e l'analisi GC/MS] è utilizzata come metodo di prova per dimostrare la conformità con i limiti di cui al primo comma.
Fino al 23 settembre 2016, si ritiene che i limiti di cui al primo comma siano rispettati se l'estratto di policiclici aromatici (PCA) è inferiore al 3 % in peso, secondo la norma dell'Institute of Petroleum IP 346:1998 (Determinazione dei PCA negli oli lubrificanti di base inutilizzati e nelle frazioni di petrolio prive di asfaltene — estrazione di dimetile solfossido), purché il rispetto dei limiti di BaP e degli elencati IPA, nonché la correlazione dei valori misurati con l'estratto PCA, siano misurati dal fabbricante o dall'importatore ogni sei mesi o dopo ogni cambio operativo di rilievo, optando per il più prossimo.
2. Inoltre, non possono essere immessi sul mercato pneumatici e battistrada per rigenerazione fabbricati dopo il 1oo gennaio 2010 se contengono oli diluenti in quantitativi superiore ai limiti fissati nel paragrafo 1.
Tali limiti sono considerati rispettati se i composti di gomma vulcanizzata non superano il limite dello 0,35 % di HBay come misurato e calcolato con il metodo ISO 21461 (gomma vulcanizzata — determinazione dell’aromaticità degli olii nei composti di gomma vulcanizzata).
3. In deroga a quanto sopra stabilito, le disposizioni del paragrafo 2 non si applicano agli pneumatici rigenerati se il loro battistrada non contiene oli diluenti che superano i limiti di cui al paragrafo 1.
4. Ai fini della presente voce, per «pneumatici» si intendono i pneumatici di veicoli contemplati nelle seguenti direttive:
- direttiva 2007/46/CE del Parlamento europeo e del Consiglio, del 5 settembre 2007, che istituisce un quadro per l’omologazione dei veicoli a motore e dei loro rimorchi (****),
- direttiva 2003/37/CE del Parlamento europeo e del Consiglio, del 26 maggio 2003, relativa all’omologazione dei trattori agricoli o forestali, dei loro rimorchi e delle loro macchine intercambiabili trainate, nonché dei sistemi, componenti ed entità tecniche di tali veicoli (*****), e
- direttiva 2002/24/CE del Parlamento europeo e del Consiglio, del 18 marzo 2002, relativa all’omologazione dei veicoli a motore a due o tre ruote e che abroga la direttiva 92/61/CEE del Consiglio (******).
5. Gli articoli non possono essere immessi in commercio per la vendita al pubblico se uno dei loro componenti in gomma o in plastica che vengono a contatto diretto e prolungato o ripetuto e a breve termine con la pelle umana o con la cavità orale, in condizioni d’ uso normali o ragionevolmente prevedibili, contiene oltre 1 mg/ kg (0,0001 % del peso di tale componente) di uno degli IPA elencati.
Tali articoli comprendono, tra l’altro:
- attrezzature sportive come le biciclette, le mazze da golf, le racchette,
- utensili per la casa, carrelli, girelli,
- attrezzi per uso domestico,
- abbigliamento, calzature, guanti e abbigliamento sportivo,
- cinturini di orologi, bracciali, maschere, fasce per i capelli.
6. I giocattoli, inclusi quelli per le attività, e gli articoli di puericultura non devono essere immessi in commercio se uno dei loro componenti che vengono a contatto diretto e prolungato oppure ripetuto e a breve termine con la pelle umana o con la cavità orale, in condizioni d’uso normali o ragionevolmente prevedibili, contiene oltre 0,5 mg/kg (0,00005 % del peso di tale componente) di uno degli IPA elencati.
7. In deroga ai paragrafi 5 e 6, la restrizione non si applica agli articoli immessi in commercio per la prima volta anteriormente al 27 dicembre 2015.
8. Entro il 27 dicembre 2017 la Commissione riesamina i valori limite di cui ai paragrafi 5 e 6 alla luce dei nuovi dati scientifici, compresi quelli relativi alla migrazione degli IPA presenti negli articoli di cui allo stesso regolamento, nonché quelli relativi a materie prime alternative e, se del caso, modifica tali paragrafi.

ID 2805 | 15.07.2016
Decreto Legislativo 22 giugno 2016 n. 128
Attuazione della direttiva 2014/53/UE concernente l’armonizzazione delle legislazioni degli Stati membri relative alla messa a disposizione sul mercato di apparecchiature radio e che abroga la direttiva 1999/5/CE.
(GU 163 del 14 Luglio 2016)
[box-warning]Modifiche al Dlgs 128/2016
- Decreto Legislativo 9 luglio 2024 n. 100 Disposizioni per l'adeguamento della normativa nazionale all'articolo 138 del regolamento (UE) 2018/1139 del Parlamento europeo e del Consiglio del 4 luglio 2018 e alla direttiva (UE) 2022/2380 del Parlamento europeo e del Consiglio del 23 novembre 2022 che modificano la direttiva 2014/53/UE, concernente l'armonizzazione delle legislazioni degli Stati membri relative alla messa a disposizione sul mercato di apparecchiature radio. (GU n.160 del 10.07.2024). Entrata in vigore: 11.07.2024[/box-warning]
Il presente decreto, in attuazione della direttiva 2014/53/UE, detta le norme per la messa a disposizione sul mercato e la messa in servizio delle apparecchiature radio.
Entrata in vigore del provvedimento: 15/07/2016
Art. 48. Disposizioni transitorie
1. È consentita la messa a disposizione sul mercato o la messa in servizio delle apparecchiature radio oggetto del presente decreto che sono conformi alla normativa vigente prima del 13 giugno 2016 e che sono state immesse sul mercato anteriormente al 13 giugno 2017.
Requisiti essenziali
Le apparecchiature radio sono fabbricate in modo da garantire:
a) la protezione della salute e della sicurezza di persone e di animali domestici e beni, compresi gli obiettivi riguardanti i requisiti di sicurezza previsti dalla direttiva 2014/35/UE e la relativa normativa di attuazione, ma senza applicazione di limiti minimi di tensione;
b) un adeguato livello di compatibilità elettromagnetica ai sensi della direttiva 2014/30/UE e la relativa normativa di attuazione.
Le apparecchiature radio sono fabbricate in modo da utilizzare efficacemente lo spettro radio e supportare l’uso efficiente dello spettro radio stesso al fine di evitare interferenze dannose.
Le apparecchiature radio di determinate categorie o classi sono fabbricate in modo tale da garantire la conformità ai seguenti requisiti essenziali:
a) interagire con accessori, in particolare con caricabatteria standardizzati;
b) interagire con altre apparecchiature radio via rete;
c) poter essere collegate a interfacce del corrispondente tipo in tutta l’Unione;
d) non danneggiare la rete o il suo funzionamento, né abusare delle risorse della rete arrecando quindi un deterioramento inaccettabile del servizio;
e) contenere elementi di salvaguardia per garantire la protezione dei dati personali e della vita privata dell’utente e dell’abbonato;
f) supportare caratteristiche speciali che consentano di tutelarsi dalle frodi;
g) supportare caratteristiche speciali che consentano l’accesso ai servizi d’emergenza;
h) supportare caratteristiche speciali che facilitino il loro uso da parte di utenti disabili;
i) supportare caratteristiche speciali che garantiscano che sia caricato un software nell’apparecchiatura radio, soltanto se è stata dimostrata la conformità della combinazione dell’apparecchiatura radio e del software.
Norme armonizzate
Le apparecchiature radio che sono conformi alle norme armonizzate o a parti di esse, i cui riferimenti sono stati pubblicati nella Gazzetta Ufficiale dell’Unione europea, sono considerate conformi ai requisiti essenziali di cui all'articolo 3, contemplati in tali norme o parti di esse.
Dichiarazione di conformità UE
La dichiarazione di conformità UE attesta il rispetto dei requisiti essenziali di cui all'articolo 3.
La dichiarazione di conformità UE ha la struttura tipo di cui all'allegato VI, contiene gli elementi indicati in tale allegato ed è continuamente aggiornata. Essa è tradotta in lingua italiana.
La dichiarazione di conformità UE semplificata di cui all'articolo 10, comma 9, contiene gli elementi di cui all'allegato VII ed è continuamente aggiornata. Essa è tradotta in lingua italiana.
Il testo integrale della dichiarazione di conformità UE è disponibile sul sito Internet indicato nella dichiarazione di conformità UE semplificata, tradotto in lingua italiana.
Se all'apparecchiatura radio si applicano più atti dell’Unione che prescrivono una dichiarazione di conformità UE, è compilata un’unica dichiarazione di conformità UE in rapporto a tutti questi atti dell’Unione. La dichiarazione contiene gli estremi degli atti dell’Unione, compresi i riferimenti della loro pubblicazione.
Con la dichiarazione di conformità UE il fabbricante si assume la responsabilità della conformità dell’apparecchiatura radio ai requisiti del presente decreto.
La documentazione tecnica contiene tutti i dati necessari o i dettagli relativi agli strumenti utilizzati dal fabbricante per garantire la conformità delle apparecchiature radio ai requisiti essenziali di cui all'articolo 3. Essa include almeno gli elementi indicati nell'allegato V.
Documentazione tecnica
La documentazione tecnica è preparata prima dell’immissione sul mercato dell’apparecchiatura radio ed è continuamente aggiornata.
La documentazione tecnica e la corrispondenza riguardanti la procedura di esame UE del tipo sono redatte in lingua italiana o in lingua inglese.
Se la documentazione tecnica non è conforme ai commi 1, 2 e 3 del presente articolo, e di conseguenza non fornisce dati o mezzi pertinenti sufficienti ad assicurare la conformità dell’apparecchiatura radio ai requisiti essenziali di cui all'articolo 3, l’autorità di sorveglianza del mercato può chiedere al fabbricante o all'importatore di far eseguire, a loro spese, una prova da un laboratorio accreditato dall'autorità di sorveglianza del mercato, ai sensi del decreto del Ministro delle comunicazioni 25 febbraio 2002, n. 84, entro un termine specifico al fine di verificare la conformità ai requisiti essenziali di cui all'articolo 3.
...
Collegati
[box-note]Nuova Direttiva R&TTE 2014/53/CE (Direttiva RED)
Dichiarazione di Conformità UE Direttiva 2016/53/UE RED
Decreto 7 aprile 2017 n. 101[/box-note]

ID 0061 | Update 17.16.2024 / Testi consolidati allegati
[box-info]Testo consolidato Ed. 27.0 del 21 Maggio 2024
Specifica Ed. 27.0 del 21 Maggio 2024:
A. Normativa
Direttiva delegata (UE) 2024/1416 della Commissione, del 13 marzo 2024, che modifica la direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda un’esenzione relativa al cadmio nei punti quantici per il downshift direttamente depositati su chip semiconduttori LED. (GU L 2024/1416 del 21.5.2024). Entrata in vigore: 10.06.2024. Applicazione dal 1° gennaio 2025.
Modifiche:
- Allegato III sostituita la voce 39 a). Inserita nota (N1)
- Allegato III aggiunta la voce 39 b). Inserita nota (N2)[/box-info]
[box-info]Testo consolidato Ed. 25.0 del 10 Gennaio 2024
Specifica Ed. 25.0 del 10.01.2024
A. Normativa
Direttiva delegata (UE) 2024/232 della Commissione, del 25 ottobre 2023, che modifica la direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa al cadmio e al piombo nei profili in plastica contenenti cloruro di polivinile rigido recuperato utilizzati per finestre e porte elettriche ed elettroniche. (GU L 2024/232 del 10.01.2024). Entrata in vigore: 30.01.2024. Applicazione a decorrere dal 1° agosto 2024.
Modifica:
- Allegato III aggiunta la voce 46. Inserita nota (N)[/box-info]
[box-info]Testo consolidato Ufficiale 01.09.2023, data di applicazione della:
- Direttiva delegata (UE) 2023/171 della Commissione del 28 ottobre 2022 che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa all’uso del cromo esavalente come agente anticorrosivo nelle pompe di calore ad assorbimento a gas. (GU L 24/33 del 26.1.2023). Applicazione del 1° settembre 2023[/box-info]
[box-info]Testo consolidato Ed. 23.0 del 24 Luglio 2023
Specifica Ed. 23.0 del 24 Luglio 2023:
A. Normativa
- Direttiva delegata (UE) 2023/1526 della Commissione del 16 maggio 2023 che modifica la direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda un’esenzione relativa al piombo come stabilizzatore termico del cloruro di polivinile impiegato come materiale di base nei sensori utilizzati nei dispositivi medico-diagnostici in vitro. (GU L 185/26 del 24.7.2023). Entrata in vigore: 13.08.2023
Modifica:
- Allegato IV punto 41 bis. Inserita nota[/box-info]
[box-info]Testo consolidato Ufficiale 01.03.2023, data di applicazione della:
- Direttiva delegata (UE) 2022/1631 della Commissione del 12 maggio 2022 che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato IV della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione riguardante l’uso del piombo sia nei cavi e nei fili superconduttori di ossido di bismuto stronzio calcio e rame sia nelle pertinenti connessioni elettriche (GU L 245/45 del 22.9.2022)
- Direttiva delegata (UE) 2022/1632 della Commissione del 12 maggio 2022 che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato IV della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa all’uso di piombo in determinati dispositivi diagnostici per la risonanza magnetica per immagini (GU L 245/48 del 22.9.2022)[/box-info]
[box-info]Testo consolidato Ufficiale 01.10.2022, data di applicazione della:
- Direttiva delegata (UE) 2021/647 della Commissione del 15 gennaio 2021 che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa all’uso di determinati composti di piombo e cromo esavalente negli iniziatori elettrici e elettronici di esplosivi per uso civile (professionale) (GU L 133 del 20.4.2021)[/box-info]
[box-info]Testo consolidato Ed. 22.0 del 12 Luglio 2023
Specifica Ed. 22.0 del 12 Luglio 2023:
A. Normativa
- Direttiva delegata (UE) 2023/1437 della Commissione del 4 maggio 2023 che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato IV della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa all’uso del mercurio nei trasduttori di pressione di fusione per i reometri capillari a determinate condizioni. (GU L 176/14 dell'11.7.2023). Applicazione: 1° febbraio 2024
Modifica:
- Allegato IV punto 49. Inserita nota[/box-info]
[box-info]Testo consolidato Ed. 21.0 del 10 Febbraio 2023
Specifica Ed. 21.0 del 10 Febbraio 2023:
A. Normativa
- Decreto Legislativo 4 marzo 2014 n. 27 (RoHS III) - Attuazione della direttiva 2011/65/UE sulla restrizione dell'uso di determinate sostanze pericolose nelle apparecchiature elettriche ed elettroniche. (GU Serie Generale n.62 del 15.03.2014). Testo consolidato 2023 con le modifiche/abrogazioni dal 2014 al 2023, in base alle modifiche disposte da:
- Decreto 16 gennaio 2023 - Modifiche all'allegato IV del decreto legislativo 4 marzo 2014, n. 27, concernente l'attuazione della direttiva 2011/65/UE sulla restrizione di determinate sostanze pericolose nelle apparecchiature elettriche ed elettroniche. (GU n.34 del 10.02.2023). Le disposizioni si applicano a decorrere dal 1° marzo 2023.[/box-info]
[box-info]Testo consolidato Ed. 20.0 del 26 Gennaio 2023
Specifica Ed. 20.0 del 26 Gennaio 2023:
A. Normativa
- Direttiva delegata (UE) 2023/171 della Commissione del 28 ottobre 2022 che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa all’uso del cromo esavalente come agente anticorrosivo nelle pompe di calore ad assorbimento a gas. (GU L 24/33 del 26.1.2023). Le disposizioni si applicano a decorrere dal 1° Settembre 2023[/box-info]
[box-info]Testo consolidato Ed. 19.0 del 22 Settembre 2022:
Specifica Ed. 19.0 del 22 Settembre 2022:
A. Normativa
- Direttiva delegata (UE) 2022/1632 della Commissione del 12 maggio 2022 che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato IV della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa all’uso di piombo in determinati dispositivi diagnostici per la risonanza magnetica per immagini. (GU L 245/48 del 22.9.2022). Le disposizioni si applicano a decorrere dal 1° marzo 2023
- Direttiva delegata (UE) 2022/1631 della Commissione del 12 maggio 2022 che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato IV della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione riguardante l’uso del piombo sia nei cavi e nei fili superconduttori di ossido di bismuto stronzio calcio e rame sia nelle pertinenti connessioni elettriche. (GU L 245/45 del 22.9.2022). Le disposizioni si applicano a decorrere dal 1° marzo 2023[/box-info]
[box-info]Testo consolidato Ed. 18.0 del 31 Agosto 2022
Specifica Ed. 18.0 del 31 Agosto 2022
A. Normativa
- Decreto Legislativo 4 marzo 2014 n. 27 (RoHS III) - Attuazione della direttiva 2011/65/UE sulla restrizione dell'uso di determinate sostanze pericolose nelle apparecchiature elettriche ed elettroniche. (GU Serie Generale n.62 del 15.03.2014). Testo consolidato 2022 con le modifiche/abrogazioni dal 2014 al 2022, in base alle modifiche disposte da:
- Decreto 4 agosto 2022 Modifiche all'allegato III del decreto legislativo 4 marzo 2014, n. 27, concernente l'attuazione della direttiva 2011/65/UE sulla restrizione dell'uso di determinate sostanze pericolose nelle apparecchiature elettriche ed elettroniche. (GU n.202 del 30.08.2022). Le disposizioni si applicano a decorrere dal 1° ottobre 2022.
Attuazione:
- direttiva delegata (UE) 2022/274,
- direttiva delegata (UE) 2022/275,
- direttiva delegata (UE) 2022/276,
- direttiva delegata (UE) 2022/277,
- direttiva delegata (UE) 2022/278,
- direttiva delegata (UE) 2022/279,
- direttiva delegata (UE) 2022/280,
- direttiva delegata (UE) 2022/281,
- direttiva delegata (UE) 2022/282,
- direttiva delegata (UE) 2022/283,
- direttiva delegata (UE) 2022/284,
- direttiva delegata (UE) 2022/287[/box-info]
[box-info]Testo consolidato Ed. 17.0 del 24 Febbraio 2022
Specifica Ed. 17.0 del 24 Febbraio 2022:
(Si applicano dal 01 Ottobre 2022)
- Direttiva delegata (UE) 2022/274 della Commissione, del 13 dicembre 2021, che modifica, adeguandolo al progresso scientifico e tecnico, l'allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l'esenzione relativa all'uso del mercurio in lampade fluorescenti a catodo freddo e lampade fluorescenti con elettrodo esterno per usi speciali (GU L 43/25 del 24.2.2022)
- Direttiva delegata (UE) 2022/275 della Commissione, del 13 dicembre 2021, che modifica, adeguandolo al progresso scientifico e tecnico, l'allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l'esenzione relativa all'uso del mercurio in altre lampade a sodio ad alta pressione (vapore) per usi generali di illuminazione (GU L 43/29 del 24.2.2022)
- Direttiva delegata (UE) 2022/276 della Commissione, del 13 dicembre 2021, che modifica, adattandolo al progresso tecnico e scientifico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda un’esenzione relativa all’uso di mercurio nelle lampade fluorescenti ad attacco singolo (compatte) per usi generali di illuminazione (GU L 43/32 del 24.2.2022)
- Direttiva delegata (UE) 2022/277 della Commissione, del 13 dicembre 2021, che modifica, adattandolo al progresso tecnico e scientifico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda un’esenzione relativa all’uso di mercurio nelle lampade fluorescenti ad attacco singolo (compatte) per usi generali di illuminazione < 30 W aventi una durata di vita di almeno 20 000 ore (GU L 43/35 del 24.2.2022)
- Direttiva delegata (UE) 2022/278 della Commissione, del 13 dicembre 2021, che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda un’esenzione relativa all’uso del mercurio nelle lampade ad alogenuri metallici (GU L 43/38 del 24.2.2022)
- Direttiva delegata (UE) 2022/279 della Commissione, del 13 dicembre 2021, che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa all’uso del mercurio in altre lampade a scarica per usi speciali (GU L 43/41 del 24.2.2022)
- Direttiva delegata (UE) 2022/280 della Commissione, del 13 dicembre 2021, che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa all’uso del mercurio in altre lampade a scarica a bassa pressione (GU L 4344 del 24.2.2022)
- Direttiva delegata (UE) 2022/281 della Commissione, del 13 dicembre 2021, che modifica, adattandolo al progresso tecnico e scientifico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda un’esenzione relativa all’uso di mercurio nelle lampade fluorescenti ad attacco singolo (compatte) per usi speciali (GU L 43/47 del 24.2.2022)
- Direttiva delegata (UE) 2022/282 della Commissione, del 13 dicembre 2021, che modifica, adattandolo al progresso tecnico e scientifico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda un’esenzione relativa all’uso di mercurio nelle lampade non lineari trifosforo (GU L 43/51 del 24.2.2022)
- Direttiva delegata (UE) 2022/283 della Commissione, del 13 dicembre 2021, che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa all’uso del mercurio in lampade a sodio ad alta pressione (vapore) con un indice di resa cromatica migliorato per usi generali di illuminazione (GU L 43/54 del 24.2.2022)
- Direttiva delegata (UE) 2022/284 della Commissione, del 16 dicembre 2021, che modifica, adeguandolo al progresso scientifico e tecnico, l’allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l’esenzione relativa all’uso del mercurio in lampade fluorescenti lineari ad attacco doppio per usi generali di illuminazione (GU L 43/57 del 24.2.2022)
- Direttiva delegata (UE) 2022/287 della Commissione, del 13 dicembre 2021, che modifica, adeguandolo al progresso scientifico e tecnico, l'allegato III della direttiva 2011/65/UE del Parlamento europeo e del Consiglio per quanto riguarda l'esenzione relativa all'uso del mercurio in lampade fluorescenti per altri usi generali di illuminazione e usi speciali (GU L 43 /64del 24.2.2022)[/box-info]
[box-info]Testo consolidato Ed. 16.0 del 16 Febbraio 2022
Specifica Ed. 16.0 del 16 Febbraio 2022:
A. Normativa
Decreto Legislativo 4 marzo 2014 n. 27 (RoHS III) - Attuazione della direttiva 2011/65/UE sulla restrizione dell'uso di determinate sostanze pericolose nelle apparecchiature elettriche ed elettroniche. (GU Serie Generale n.62 del 15-03-2014). Testo consolidato 2022 con le modifiche/abrogazioni dal 2014 al 2022, in base alle modifiche disposte da:
- Decreto 28 dicembre 2021 Attuazione delle direttive delegate della Commissione europea (UE) 2021/1978, (UE) 2021/1979 e (UE) 2021/1980, dell'11 agosto 2021, di modifica dell'allegato IV del decreto legislativo 4 marzo 2014, n. 27, sulla restrizione di determinate sostanze pericolose nelle apparecchiature elettriche ed elettroniche (ROHS II), in GU n. 38 del 15.02.2022.
Le disposizioni si applicano a decorrere dal 21 luglio 2021.[/box-info]
[box-warning]RoHS III
Con la Direttiva delegata (UE) 2015/863, (si applica dal 22 luglio 2019) si è giunti alla III integrazione della restrizione dell’uso di determinate sostanze pericolose nelle apparecchiature elettriche ed elettroniche (RoHS III).[/box-warning]
Vedi il testo consolidato 2023
Direttiva 2011/65/UE del Parlamento e del Consiglio dell’8 giugno 2011 sulla restrizione dell’uso di determinate sostanze pericolose nelle apparecchiature elettriche ed elettroniche - RoHS II
GU L 174 del 1.7.2011
______
La Direttiva 2011/65/CE (RoHS 2) istituisce norme riguardanti la restrizione all’uso di sostanze pericolose nelle apparecchiature elettriche ed elettroniche (AEE) al fine di contribuire alla tutela della salute umana e dell’ambiente, compresi il recupero e lo smaltimento ecologicamente corretti dei rifiuti di AEE.
Rispetto alla RoHS 1 (Direttiva 2002/95/CE) la RoHS2 si presenta come una direttiva a sé stante (e non più interconnessa alla Diretiva RAEE): rispetto alla precedente formulazione sono stati eliminati i collegamenti alla RAEE, ad esempio per quanto riguarda l’elenco delle apparecchiature incluse nel campo di applicazione, il quale viene invece definito all’allegato I della nuova direttiva.
Il campo di applicazione della nuova direttiva è sostanzialmente ampliato con l’aggiunta di una nuova categoria che include tutte le AEE non coperte da alcuna delle altre 10 categorie.
Categorie di AEE disciplinate dalla RoHS 2:
- Grandi elettrodomestici
- Piccoli elettrodomestici
- Apparecchiature informatiche e per telecomunicazioni
- Apparecchiature di consumo
- Apparecchiature di illuminazione
- Strumenti elettrici ed elettronici
- Giocattoli e apparecchiature per il tempo libero e per lo sport
- Dispositivi medici
- Strumenti di monitoraggio e controllo, compresi gli strumenti di monitoraggio e controllo industriali
- Distributori automatici
- Altre AEE non comprese nelle categorie sopra elencate (open scope)
Sostanze con restrizioni d’uso e valori delle concentrazioni massime tollerate per peso nei materiali omogenei:
- Piombo (0,1 %)
- Mercurio (0,1 %)
- Cadmio (0,01 %)
- Cromo esavalente (0,1 %)
- Bifenili polibromurati (PBB) (0,1 %)
- Eteri di difenile polibromurato (PBDE) (0,1 %)
- Ftalato di bis(2-etilesile) (DEHP) (0,1%) | Aggiunta da Direttiva delegata (UE) 2015/863 | ROHS III
- Benzilbutilftalato (BBP) (0,1%) | Aggiunta da Direttiva Delegata delegata (UE) 2015/863 | ROHS III
- Dibutilftalato (DBP) (0,1%) | Aggiunta da Direttiva delegata (UE) 2015/863 | ROHS III
- Diisobutilftalato (DIBP) (0,1%) | New RoHS III | Aggiunta da Direttiva delegata (UE) 2015/863 | ROHS III
Definizioni:
1) «apparecchiature elettriche ed elettroniche» o «AEE», le apparecchiature che dipendono, per un corretto funzionamento, da correnti elettriche o campi elettromagnetici e le apparecchiature di generazione, trasferimento e misura di tali correnti e campi e progettate per essere usate con una tensione non superiore a 1 000 volt per la corrente alternata e a 1 500 volt per la corrente continua;
2) ai fini del punto 1, «che dipendono», in relazione alle AEE, indica il fatto che le apparecchiature necessitano di correnti elettriche o di campi elettromagnetici per espletare almeno una delle funzioni previste;
3) «utensili industriali fissi di grandi dimensioni», un insieme di grandi dimensioni di macchine, apparecchiature e/o componenti, che funzionano congiuntamente per un’applicazione specifica, installati e disinstallati in maniera permanente da professionisti in un determinato luogo e utilizzati e gestiti da professionisti presso un impianto di produzione industriale o un centro di ricerca e sviluppo;
4) «impianto fisso di grandi dimensioni», una combinazione su larga scala di apparecchi di vario tipo ed eventualmente di altri dispositivi, che sono assemblati e installati da professionisti, destinati ad essere utilizzati in modo permanente in un luogo prestabilito e apposito e disinstallati da professionisti;
5) «cavi», tutti i cavi con una tensione nominale inferiore ai 250 volt che servono da collegamento o da prolunga per collegare le AEE alla presa elettrica o per collegare tra di loro una o più AEE;
6) «fabbricante», qualsiasi persona fisica o giuridica che fabbrica un’AEE, oppure che la fa progettare o fabbricare e la commercializza apponendovi il proprio nome o marchio;
7) «mandatario», qualsiasi persona fisica o giuridica stabilita nell’Unione che abbia ricevuto da un fabbricante un mandato scritto che la autorizza ad agire per suo conto in relazione a determinate attività;
8) «distributore», qualsiasi persona fisica o giuridica nella catena di fornitura, diversa dal fabbricante o dall’importatore, che mette a disposizione un’AEE sul mercato;
9) «importatore», qualsiasi persona fisica o giuridica stabilita nell’Unione che immetta sul mercato dell’Unione un’AEE originaria di un paese terzo;
10) «operatori economici», il fabbricante, il mandatario, l’importatore e il distributore;
11) «messa a disposizione sul mercato», qualsiasi fornitura di un’AEE per la distribuzione, il consumo o l’uso sul mercato dell’Unione nel corso di un’attività commerciale, a titolo oneroso o gratuito;
12) «immissione sul mercato», la prima messa a disposizione di un’AEE sul mercato dell’Unione;
13) «norma armonizzata», una norma adottata da uno degli organismi europei di normalizzazione elencati nell’allegato I della direttiva 98/34/CE del Parlamento europeo e del Consiglio, del 22 giugno 1998, che prevede una procedura d’informazione nel settore delle norme e delle regolamentazioni tecniche e delle regole relative ai servizi della società dell’informazione (1), sulla base di una richiesta presentata dalla Commissione conformemente all’articolo 6 di tale direttiva;
14) «specificazione tecnica», un documento che prescrive i requisiti tecnici che un prodotto, un processo o un servizio devono soddisfare;
15) «marcatura CE», una marcatura mediante cui il fabbricante indica che il prodotto è conforme ai requisiti applicabili stabiliti dalla normativa dell’Unione di armonizzazione che ne prevede l’apposizione;
16) «valutazione della conformità», la procedura atta a dimostrare se le prescrizioni della presente direttiva in materia di AEE siano state rispettate;
17) «vigilanza del mercato», le attività svolte e i provvedimenti adottati dalle autorità pubbliche per garantire che le AEE siano conformi ai requisiti stabiliti nella presente direttiva e non pregiudichino la salute, la sicurezza o qualsiasi altro aspetto della tutela del pubblico interesse;
18) «richiamo», qualsiasi provvedimento volto ad ottenere la restituzione di un prodotto che è già stato messo a disposizione dell’utilizzatore finale;
19) «ritiro», qualsiasi provvedimento volto a impedire la messa a disposizione sul mercato di un prodotto nella catena di fornitura;
20) «materiale omogeneo», un materiale di composizione uniforme o un materiale costituito dalla combinazione di più materiali che non può essere diviso o separato in materiali diversi mediante azioni meccaniche come lo svitamento, il taglio, la frantumazione, la molatura e processi abrasivi;
21) «dispositivo medico», un dispositivo medico ai sensi dell’articolo 1, paragrafo 2, lettera a), della direttiva 93/42/CEE e che è anche un’AEE;
22) «dispositivo medico-diagnostico in vitro», un dispositivo medico-diagnostico in vitro ai sensi dell’articolo 1, paragrafo 2, lettera b), della direttiva 98/79/CE;
23) «dispositivo medico impiantabile attivo», qualsiasi dispositivo medico impiantabile attivo ai sensi dell’articolo 1, paragrafo 2, lettera c), della direttiva 90/385/CEE del Consiglio, del 20 giugno 1990, per il ravvicinamento delle legislazioni degli Stati membri relative ai dispositivi medici impiantabili attivi (2);
24) «strumenti di monitoraggio e controllo industriali», strumenti di monitoraggio e controllo destinati esclusivamente ad uso industriale o professionale;
25) «disponibilità di un sostituto», la capacità di un sostituto di essere fabbricato e consegnato entro un ragionevole lasso di tempo rispetto al tempo necessario per la fabbricazione e la dis-tribuzione delle sostanze di cui all’allegato II;
26) «affidabilità di un sostituto», la probabilità che un’AEE che utilizza un sostituto esegua una funzione richiesta senza guasti, in determinate condizioni, per un determinato periodo di tempo;
27) «pezzo di ricambio», una parte distinta di un’AEE che può sostituire una parte di un’AEE. L’AEE non può funzionare come previsto in assenza di tale parte. La funzionalità dell’AEE è ristabilita o è potenziata quando la parte è sostituita da un pezzo di ricambio;
28) «macchine mobili non stradali destinate ad esclusivo uso professionale», le macchine dotate di una fonte di alimentazione a bordo il cui funzionamento richiede mobilità o movimento continuo o semicontinuo, durante il lavoro, tra una serie di postazioni di lavoro fisse e sono destinate ad esclusivo uso professionale.
Modifiche:
M1 - Direttiva delegata 2012/50/UE della Commissione del 10 ottobre 2012
M2 - Direttiva delegata 2012/51/UE della Commissione del 10 ottobre 2012
M3 - Direttiva delegata 2014/1/UE della Commissione, del 18 ottobre 2013
M4 - Direttiva delegata 2014/2/UE della Commissione, del 18 ottobre 2013
M5 - Direttiva delegata 2014/3/UE della Commissione, del 18 ottobre 2013
M6 - Direttiva delegata 2014/4/UE della Commissione, del 18 ottobre 2013
M7 - Direttiva delegata 2014/5/UE della Commissione, del 18 ottobre 2013
M8 - Direttiva delegata 2014/6/UE della Commissione, del 18 ottobre 2013
M9 - Direttiva delegata 2014/7/UE della Commissione, del 18 ottobre 2013
M10 - Direttiva delegata 2014/8/UE della Commissione, del 18 ottobre 2013
M11 - Direttiva delegata 2014/9/UE della Commissione, del 18 ottobre 2013
M12 - Direttiva delegata 2014/10/UE della Commissione, del 18 ottobre 2013
M13 - Direttiva delegata 2014/11/UE della Commissione, del 18 ottobre 2013
M14 - Direttiva delegata 2014/12/UE della Commissione, del 18 ottobre 2013
M15 - Direttiva delegata 2014/13/UE della Commissione, del 18 ottobre 2013
M16 - Direttiva delegata 2014/14/UE della Commissione, del 18 ottobre 2013
M17 - Direttiva delegata 2014/15/UE della Commissione, del 18 ottobre 2013
M18 - Direttiva delegata 2014/16/UE della Commissione, del 18 ottobre 2013
M19 - Direttiva delegata 2014/69/UE della Commissione, del 13 marzo 2014
M20 - Direttiva delegata 2014/70/UE della Commissione, del 13 marzo 2014
M21 - Direttiva delegata 2014/71/UE della Commissione, del 13 marzo 2014
M22 - Direttiva delegata 2014/72/UE della Commissione, del 13 marzo 2014
M23 - Direttiva delegata 2014/73/UE della Commissione, del 13 marzo 2014
M24 - Direttiva delegata 2014/74/UE della Commissione, del 13 marzo 2014
M25 - Direttiva delegata 2014/75/UE della Commissione, del 13 marzo 2014
M26 - Direttiva delegata 2014/76/UE della Commissione, del 13 marzo 2014
M27 - Direttiva delegata (UE) 2015/573 della Commissione del 30 gennaio 2015
M28 - Direttiva delegata (UE) 2015/574 della Commissione del 30 gennaio 2015
M29 - Direttiva delegata (UE) 2015/863 della Commissione del 31 marzo 2015 (GU L 137/11 4.6.2015) - Si applica dal: 22 luglio 2019 - RoHS III (modifica All. II)
M30 - Direttiva delegata (UE) 2016/585 della Commissione del 12 febbraio 2016 (GU L 101 12 16.4.2016)
M31 - Direttiva delegata (UE) 2016/1028 della Commissione del 19 aprile 2016 (GU L 168 del 25.6.2016) - Si applica dal: 20 luglio 2016
M32 - Direttiva delegata (UE) 2016/1029 della Commissione del 19 aprile 2016 (GU L 168 del 25.6.2016) - Si applica dal: 20 luglio 2016
M33 - Direttiva delegata (UE) 2017/1009 della Commissione del 13 marzo 2017 (GU L 153/21 del 16.6.2017) - Si applica dal: 6 luglio 2018
M34 - Direttiva delegata (UE) 2017/1010 della Commissione del 13 marzo 2017 (GU L 153/23 del 16.06.2017) - Si applica dal: 6 luglio 2018
M35 - Direttiva delegata (UE) 2017/1011 della Commissione del 15 marzo 2017 (GU L 153/25 del 16.06.2017) - Si applica dal: 6 luglio 2018
M36 - Direttiva delegata (UE) 2017/1975 della Commissione del 7 agosto 2017 (GU L 281/29 del 31.10.2017)
M37 - Direttiva (UE) 2017/2102 del Parlamento Europeo e del Consiglio del 15 novembre 2017 (GU L 305/08 del 21.11.2017) Si applica dal: 22 luglio 2019
M38 - Direttiva delegata (UE) 2018/736 della Commissione del 27 febbraio 2018 - in applicazione dal 1° luglio 2019.
M39 - Direttiva delegata (UE) 2018/737 della Commissione del 27 febbraio 2018 - in applicazione dal 1° luglio 2019.
M40 - Direttiva delegata (UE) 2018/738 della Commissione del 27 febbraio 2018 - in applicazione dal 1° luglio 2019.
M41 - Direttiva delegata (UE) 2018/739 della Commissione del 1° marzo 2018 - in applicazione dal 1° luglio 2019.
M42 - Direttiva delegata (UE) 2018/740 della Commissione del 1° marzo 2018 - in applicazione dal 1° luglio 2019.
M43 - Direttiva delegata (UE) 2018/741 della Commissione del 1° marzo 2018 - in applicazione dal 1° luglio 2019.
M44 - Direttiva delegata (UE) 2018/742 della Commissione del 1° marzo 2018 - in applicazione dal 1° luglio 2019.
M45 - Direttiva delegata (UE) 2019/169 della Commissione del 16 novembre 2018 - in applicazione dal 1° marzo 2020.
M46 - Direttiva delegata (UE) 2019/170 della Commissione del 16 novembre 2018 - in applicazione dal 1° marzo 2020.
M47 - Direttiva delegata (UE) 2019/171 della Commissione del 16 novembre 2018 - in applicazione dal 1° marzo 2020.
M48 - Direttiva delegata (UE) 2019/172 della Commissione del 16 novembre 2018 - in applicazione dal 1° marzo 2020.
M49 - Direttiva delegata (UE) 2019/173 della Commissione del 16 novembre 2018 - in applicazione dal 1° marzo 2020.
M50 - Direttiva delegata (UE) 2019/174 della Commissione del 16 novembre 2018 - in applicazione dal 1° marzo 2020.
M51 - Direttiva delegata (UE) 2019/175 della Commissione del 16 novembre 2018 - in applicazione dal 1° marzo 2020.
M52 - Direttiva delegata (UE) 2019/176 della Commissione del 16 novembre 2018 - in applicazione dal 1° marzo 2020.
M53 - Direttiva delegata (UE) 2019/177 della Commissione del 16 novembre 2018 - in applicazione dal 1° marzo 2020.
M54 - Direttiva delegata (UE) 2019/178 della Commissione del 16 novembre 2018 - in applicazione dal 1° marzo 2020.
M55 - Direttiva delegata (UE) 2019/1845 della Commissione dell’8 agosto 2019 - in applicazione dal 1°maggio 2020.
M56 - Direttiva delegata (UE) 2019/1846 della Commissione dell’8 agosto 2019 - in applicazione dal 1°maggio 2020.
M57 - Direttiva delegata (UE) 2020/360 della Commissione del 17 dicembre 2019 - in applicazione dal 1° aprile 2021.
M58 - Direttiva delegata (UE) 2020/361 della Commissione del 17 dicembre 2019 - in applicazione dal 1° aprile 2021.
M59 - Direttiva delegata (UE) 2020/364 della Commissione del 17 dicembre 2019 - in applicazione dal 1° settembre 2020.
M60 - Direttiva delegata (UE) 2020/365 della Commissione del 17 dicembre 2019 - in applicazione dal 1° aprile 2021.
M61 - Direttiva delegata (UE) 2020/366 della Commissione del 17 dicembre 2019 - in applicazione dal 1° aprile 2021.
M62 - Direttiva delegata (UE) 2021/647 della Commissione del 15 gennaio 2021 - in applicazione dal 1° novembre 2021. - Testo consolidato 01.11.2021
M63 - Direttiva delegata (UE) 2021/884 della Commissione dell’8 marzo 2021 - in applicazione dal 1° luglio 2022.
M64 - Direttiva delegata (UE) 2021/1978 della Commissione dell'11 agosto 2021 - in applicazione dal 21 Luglio 2021.
M65 - Direttiva delegata (UE) 2021/1979 della Commissione dell'11 agosto 2021 - in applicazione dal 21 Luglio 2021.
M66 - Direttiva delegata (UE) 2021/1980 della Commissione dell'11 agosto 2021 - in applicazione dal 21 Luglio 2021. - Testo consolidato 01.07.2022
M67 - Direttiva delegata (UE) 2022/274 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M68 - Direttiva delegata (UE) 2022/275 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M69 - Direttiva delegata (UE) 2022/276 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M70 - Direttiva delegata (UE) 2022/277 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M71 - Direttiva delegata (UE) 2022/278 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M72 - Direttiva delegata (UE) 2022/279 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M73 - Direttiva delegata (UE) 2022/280 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M74 - Direttiva delegata (UE) 2022/281 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M75 - Direttiva delegata (UE) 2022/282 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M76 - Direttiva delegata (UE) 2022/283 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M77 - Direttiva delegata (UE) 2022/284 della Commissione, del 16 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M78 - Direttiva delegata (UE) 2022/287 della Commissione, del 13 dicembre 2021 - in applicazione dal 1° Ottobre 2022. - Testo consolidato 01.10.2022
M79 - Direttiva delegata (UE) 2022/1631 della Commissione del 12 maggio 2022 - in applicazione dal 1° Marzo 2023. - Testo consolidato 01.03.2023
M80 - Direttiva delegata (UE) 2022/1632 della Commissione del 12 maggio 2022 - in applicazione dal 1° Marzo 2023. - Testo consolidato 01.03.2023
M81 - Direttiva delegata (UE) 2023/171 della Commissione del 28 ottobre 2022 - in applicazione dal 1° Settembre 2023 - Testo consolidato 01.09.2023
M82 - Direttiva delegata (UE) 2023/1437 della Commissione del 4 maggio 2023 - in applicazione dal 1° Febbraio 2024 - Testo consolidato 01.02.2024
M83 - Direttiva delegata (UE) 2023/1526 della Commissione del 16 maggio 2023 (GU L 185/26 del 24.7.2023) - Testo consolidato 01.02.2024
M84 - Direttiva delegata (UE) 2024/232 della Commissione del 25 ottobre 2023 - in applicazione dal 1° Agosto 2024 - Testo consolidato 01.08.2024
--- - Direttiva delegata (UE) 2024/1416 della Commissione del 13 marzo 2024 - in applicazione dal 1° Gennaio 2025 [...]
Rettifiche:
- Rettifica della direttiva 2011/65/UE del Parlamento europeo e del Consiglio, dell’8 giugno 2011
- Rettifica della direttiva delegata (UE) 2017/1975 della Commissione del 7 agosto 2017 (GU L 285/32 del 01.11.2017)
- Rettifica della direttiva 2011/65/UE del Parlamento europeo e del Consiglio, dell’8 giugno 2011 (GU L 268/77 del 22.10.2019)
...

Vedi il Testo consolidato RoHS
Collegati
[box-note]Direttiva RoHS: Open Scope dal 22 luglio 2019
Direttiva RoHS: Evoluzioni
Direttiva (UE) 2015/863: Modifica all'allegato II RoHS II
Dichiarazione UE di Conformità RoHS II - Modello
Direttiva RoHS 2 Testo Consolidato 2017
Decreto Legislativo n. 27 del 4 Marzo 2014
Indicazioni operative ambito applicazione direttiva RAEE[/box-note]

Direttiva 2008/57/CE del Parlamento europeo e del Consiglio del 17 giugno 2008 relativa all’interoperabilità del sistema ferroviario comunitario
GUUE L191/15 del 18.07.2008
Entrata in vigore: 07.08.2008
[box-warning]Abrogata da:
Direttiva (UE) 2016/797
Direttiva (UE) 2016/798[/box-warning]
Normativa di attuazione:
[box-note]Decreto Legislativo 8 ottobre 2010 n.191[/box-note]

The European Parliament and the Council have reached an agreement for better surveillance and traceability of medical and in vitro diagnostic devices.
Medical devices are medical equipment and devices for in vitro tests used either in hospitals by health professionals or directly by the patient.
There are over 500,000 types of medical devices and devices for in vitro tests on the EU market. These cover a wide range of products from home-use items like sticking plasters, pregnancy tests and contact lenses to x-ray machines, pacemakers, breast implants, hip replacements and HIV blood tests.
This new agreement between the European Parliament and the Council from 15 June 2016 takes on board some key concerns of the Commission indicated at several stages of the negotiation and contains a series of important improvements to the current system:
The regulations establish a modernised and more robust EU legislative framework that is crucial to ensure a better protection of public health and patient safety.
They will also contribute to innovation in this sector, to the competitiveness of the EU industry and to the better functioning of the Internal Market in this strategic sector.
Next steps
The Council has planned to approve the agreement at ministers' level in September. Following their legal-linguistic review, the two draft regulations will be then adopted by the Parliament and the Council, probably at the end of the year or early 2017.
The new rules will apply 3 years after publication for medical devices and 5 years after publication for in vitro diagnostic medical devices.
Background
Medical devices are currently regulated by 3 main Directives:
The existing regulatory framework has proved its merits, but weaknesses have highlighted the need for revision. In addition, following scandals involving medical devices in recent years, the current legislative framework has come under harsh criticism in the media and on the political arena.
It is in this context that, on 26 September 2012, the European Commission presented two legislative proposals to ensure that medical devices in the EU are safe, effective and innovative.
Over the past years, the Commission has been working hard with the European Parliament and the Council to facilitate the adoption of these legislative proposals as soon as possible.
The medical devices sector is a major employer in Europe: it employs more than 575,000 people in over 22,500 companies and has an annual turnover of €110 billion. The sector is driven by small and medium-sized companies, which make up almost 95% of the industry.
About 6-8% of medical devices' annual sales and 10% of in vitro devices' annual sales is re-invested in research every year.
Report 24 del 17/06/2016
N.23 A12/0743/16 Lettonia
Approfondimento tecnico: Estintore


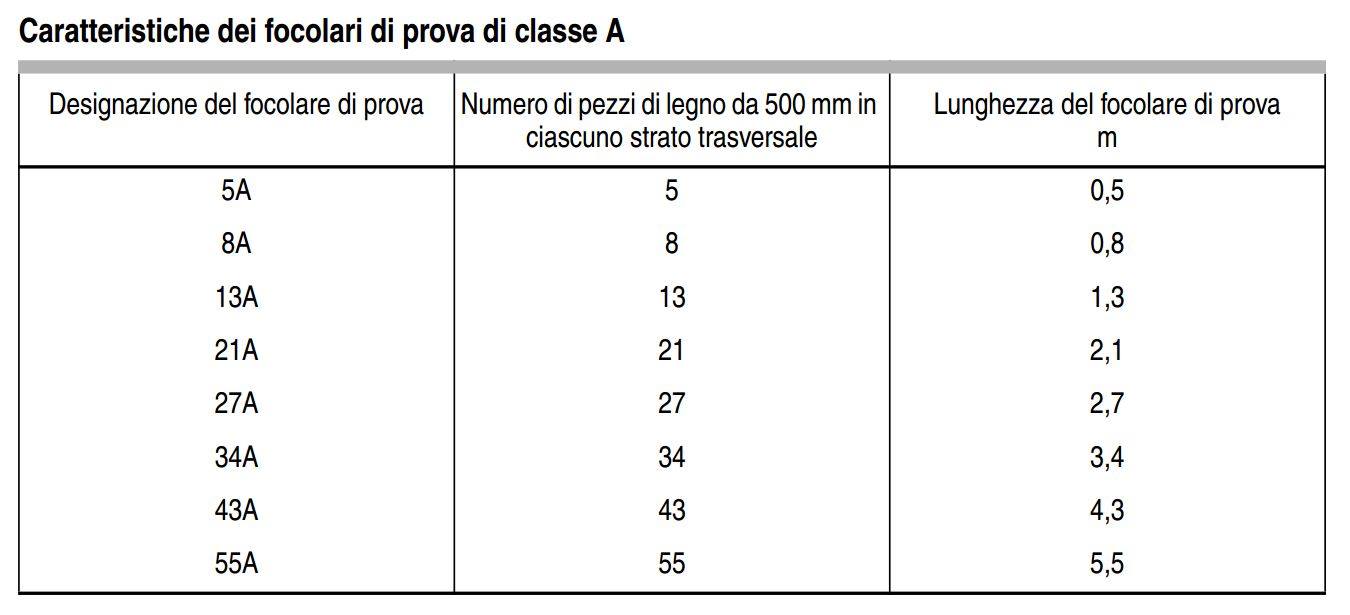
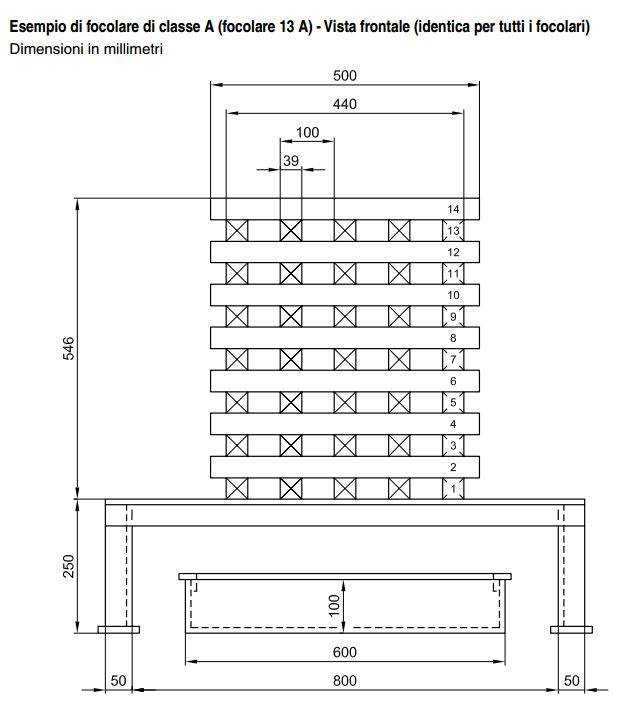
Il prodotto, di marca VADER, Modello 1309 1 Kg, è stato distrutto dal Fabbricante perché non conforme alla Direttiva 97/23/CE del Parlamento e Europeo del Consiglio del 29 maggio 1997 per il ravvicinamento delle legislazioni degli Stati membri in materia di attrezzature a pressione ed alla norma tecnica EN 3-7.
In caso di incendio, l’estintore non spegne in modo efficiente le fiamme sottoponendo l’utilizzatore al rischio di ustione.
Tutti gli estintori per poter essere “classificati” devono essere sottoposti alle prove di spegnimento previste dall’Appendice I della norma EN 3-7.
L’appendice I descrive come effettuare le prove con focolari di classe A e con focolari di classe B.
Caratteristiche dei focolari di prova di classe A.
I focolari di prova di classe A devono essere costituiti da una pila di legna sostenuta da un’intelaiatura metallica di 250 mm di altezza, 900 mm di larghezza e lunghezza pari a quella del focolare di prova. L’intelaiatura metallica deve essere costituita da profilati angolari (L × L) (50 ⋅ 50) mm come specificato nella ISO 657-1.
Ciascun focolare di prova è contraddistinto da un numero (che indica le dimensioni del focolare) seguito dalla lettera A.
Caratteristiche dei focolari di prova di classe B.
I focolari di prova di classe B devono essere realizzati in una serie di piatti circolari di lamiera d’acciaio saldata. Lo spessore nominale della base deve essere lo stesso delle pareti e la tolleranza per lo spessore del materiale della base e delle pareti deve essere conforme alla norma nazionale applicabile.
Barre o profilati di rinforzo possono essere saldati al lato inferiore della base, con una distanza minima di 200 mm tra gli elementi di rinforzo sostanzialmente paralleli. Tutte le tolleranze specificate si riferiscono al piatto al momento della fabbricazione.
I piatti devono contenere acqua ricoperta da uno strato di combustibile nelle proporzioni seguenti: ¹⁄₃ di acqua, ²⁄₃ di combustibile. Il volume totale del liquido nel piatto deve essere corrispondente a una profondità dell’acqua di circa 10 mm e a una profondità del combustibile di circa 20 mm.
I focolari di prova sono contraddistinti da un numero (che indica le dimensioni del focolare) seguito dalla lettera B.
A seguito delle prove si classificano le capacità estinguenti degli estintori.
Report 23 del 10/06/2016
N.11 A12/0721/16 Francia
Approfondimento tecnico: Crema per la pelle


Il prodotto, di marca Dream Cosmetics, è stato sottoposto alla procedura di ritiro dal mercato perché non conforme al REGOLAMENTO (CE) n. 1223/2009 DEL PARLAMENTO EUROPEO E DEL CONSIGLIO del 30 novembre 2009 sui prodotti cosmetici.
La crema contiene clobetasolo propionato (concentrazione misurata uguale a 0,04% in peso). Il clobetasolo è un corticosteroide da applicare sulla pelle solo se prescritto per la cura di alcune malattie della stessa. Oltre a schiarire la pelle, può provocare eruzioni cutanee e bruciori nella zona di applicazione ed, a lungo termine, potrebbe causare problemi all’apparato endocrino.
Il REGOLAMENTO (CE) n. 1223/2009, all’art. 14, stabilisce che:
1. Fatto salvo l’articolo 3, i prodotti cosmetici non possono contenere:
a) sostanze vietate:
- sostanze vietate di cui all'allegato II;
b) sostanze soggette a restrizioni:
- sostanze soggette a restrizioni non impiegate conformemente alle restrizioni indicate nell'allegato III;
c) coloranti:
i) coloranti diversi da quelli elencati nell’allegato IV e coloranti ivi elencati ma non impiegati conformemente alle condizioni indicate nel suddetto allegato, ad eccezione dei prodotti per la colorazione dei capelli di cui al paragrafo 2;
ii) fatte salve la lettera b), la lettera d), punto i) e la lettera
e), punto i), sostanze elencate nell’allegato IV ma non destinate ad essere impiegate come coloranti, e non impiegate conformemente alle condizioni indicate nel suddetto allegato.
d) conservanti:
i) conservanti diversi da quelli elencati nell’allegato V e conservanti ivi elencati ma non impiegati conformemente alle condizioni indicate nel suddetto allegato;
ii) fatte salve la lettera b, la lettera c), punto i) e la lettera e), punto
i), sostanze elencate nell’allegato V ma non destinate ad essere impiegate come conservanti, e non impiegate conformemente alle condizioni indicate nel suddetto allegato.
e) filtri UV:
i) Filtri UV diversi da quelli elencati nell’Allegato VI e filtri UV ivi elencati ma non impiegati conformemente alle condizioni indicate nel suddetto allegato;
ii) fatte salve la lettera b), la lettera c), punto i), e la lettera
d), punto i), sostanze elencate nell’allegato VI ma non destinate ad essere impiegate come filtri UV, e non impiegate conformemente alle condizioni indicate nel suddetto allegato.
I corticosteroidi fanno parte delle sostanze elencate nell’Allegato II del REGOLAMENTO (CE) n. 1223/2009.

Update Ed. 2.0 Luglio 2017
Aggiornata la raccolta segnaletica ISO:2011 (UNI 2012) alla versione ISO 2016 (UNI 2017), con altri segnali inseriti nell'emendamento 7
- Prospetto della codifica dei segnaletica
- Raccolta immagini segnaletica
_______
Ed. 1.0 2015
Aggiornata la raccolta segnaletica ISO:2011 (UNI 2012) alla versione ISO 2014 (UNI 2015 Assente emendamento ISO 6:2014), quindi da 6 emendamenti che introducono altri 95 cartelli.
ISO 7010:2011
Graphical symbols - Safety colours and safety signs - Registered safety signs
ISO 7010:2011/Amd 1:2012
ISO 7010:2011/Amd 2:2012
ISO 7010:2011/Amd 3:2012
ISO 7010:2011/Amd 4:2013
ISO 7010:2011/Amd 5:2014
ISO 7010:2011/Amd 6:2014
La norma prescrive i segnali di sicurezza da utilizzare nella prevenzione degli infortuni, nella protezione dal fuoco, per l’informazione sui pericoli alla salute e nelle evacuazioni di emergenza.
La forma e il colore di ogni segnale di sicurezza sono conformi alla ISO 3864-1 e la progettazione dei segni grafici è conforme alla ISO 3864-3.
La norma è applicabile a tutti i siti in cui le questioni legate alla sicurezza delle persone necessitano di essere poste.Comunque, non è applicabile ai segnali utilizzati nel traffico ferroviario, stradale, fluviale, marittimo e aereo e, in generale, in quei settori soggetti a una regolamentazione che può differire in alcuni punti della presente norma e della serie ISO 3864.
La norma specifica gli originali dei segnali di sicurezza che possono essere ridotti o ingranditi per esigenze di riproduzione e di applicazione.
Fonte ISO:
http://www.iso.org/iso/home/search.htm?qt=7010&sort=rel&type=simple&published=onNumero: 206
Fonte UNI:
http://store.uni.com/magento-1.4.0.1/index.php/uni-en-iso-7010-2015.html
Dim.: 800 x 800 px
Formato: png
Raccolta Elaborata su norma Licenza ISO/EVS
[box-note]Raccolte pubblicate:
ISO 7010 Raccolta segnaletica di sicurezza - Ed. 2021
ISO 7010 Raccolta segnaletica di sicurezza - Ed. 2020
ISO 7010 Raccolta Segnaletica sicurezza - Ed. 2018
ISO 7010 Raccolta Segnaletica sicurezza - Ed. 2017
La Raccolta dei Segnali di sicurezza EN ISO Ed. 2012[/box-note]
Maggiori Info e acquisto Documento
Documento compreso nel Servizio di Abbonamento Tema Marcatura CE
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| ISO 7010 + Amendment List of Signs.pdf Lista della segnaletica ISO 7010 Ed. 2015 |
256 kB | 472 | ||
| ISO 7010 + AMENDMENT.zip Raccolta segnaletica Ed. 2015 |
15450 kB | 226 |
Draft guidance document on the ElectroMagnetic Compatibility Directive transition from 2004/108/EC to 2014/30/EU.
The new Electromagnetic Compatibility Directive 2014/30/EU(1) is the result of the alignment of the previous Electromagnetic Compatibility Directive 2004/108/EC to the "New Legislative Framework" (NLF), in particular to Decision No 768/2008/EC3, as well as to the provisions of the Treaty on the Functioning of the European Union (TFEU) after the Treaty of Lisbon.
Being the result of an alignment and a recast, the main changes in the new Directive 2014/30/EU with respect to the previous Directive 2004/108/EC are quite limited, and do not concern the most substantial characteristics of the act that remain the same: scope, essential requirements and conformity assessment procedure.
The main changes are the following:
- Reference number: according to the model YYYY /No/UE
- Definitions: horizontal additions from the NLF
- Economic operators (manufacturers, authorised representatives, importers, distributors) and their obligations: more detailed descriptions from the NLF
- Harmonised standards and presumption of conformity: reference to Regulation (EU) No 1025/2012 on European Standardisation(4)
- CE marking: reference to Regulation (EC) No 765/2008(5)
- Notified bodies: interpretation of the requirements and procedures of Decision 768/2008 in the context of EMC.
- Market surveillance and safeguard procedure: reinforced activities and new simplified procedures (also related to the "Product safety and market surveillance package"(6) )
- Committee on Electromagnetic Compatibility and implementing acts: reference to Regulation (EU) No 182/20117 ("Comitology") concerning Commission Implementing Decisions on formal objections against harmonised standards and safeguard clauses against products
- EU declaration of conformity: more detailed contents, and a model, from Decision 768/2008
- EU-type examination certificate: conditions on validity and date of expiry from the NLF
The new Electromagnetic Compatibility Directive 2014/30/EU is applicable from 20 April 2016.
This document includes a list of "Frequently Asked Questions and Answers" on the transition to the Electromagnetic Compatibility Directive 2014/30/EU, which covers both "horizontal" and "sectorial" questions, this is to say, those common to all the EU legislation aligned to the "New Legislative Framework"8 and those specifically related to Directive 2014/30/EU.
It reflects the result of ongoing discussions, notably at the workshop on the transition to the new EMCD 2014/30/EU held on 12 November 2014.
European Commission 2016
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| Guidance EMC Transition Directive 2004 108 CE to 2014 30 UE.pdf European Commission 2016 |
526 kB | 129 |
Report 32 del 12/08/2016 N.7 A12/0968/16 Portogallo
Approfondimento tecnico: Illuminante per la pelle

Il prodotto, di marca “CaroWhite”, è stato sottoposto alla procedura di blocco dell’importazione perché non conforme con il Regolamento (CE) n. 1223/2009 del Parlamento europeo e del Consiglio del 30 novembre 2009, sui prodotti cosmetici.
Il prodotto contiene idrochinone, che può causare irritazioni cutanee e dermatiti.
Il Regolamento (CE) n. 1223/2009 stabilisce che:
CAPO IV
RESTRIZIONI APPLICABILI A DETERMINATE SOSTANZE
Articolo 14
Restrizioni applicabili alle sostanze elencate negli allegati
1. Fatto salvo l’articolo 3, i prodotti cosmetici non possono contenere:
a) sostanze vietate:
- sostanze vietate di cui all’allegato II;
b) sostanze soggette a restrizioni:
- sostanze soggette a restrizioni non impiegate conformemente alle restrizioni indicate nell’allegato III;
c) coloranti:
i) coloranti diversi da quelli elencati nell’allegato IV e coloranti ivi elencati ma non impiegati conformemente alle condizioni indicate nel suddetto allegato, ad eccezione dei prodotti per la colorazione dei capelli di cui al paragrafo 2;
ii) fatte salve la lettera b), la lettera d), punto i) e la lettera e), punto i), sostanze elencate nell’allegato IV ma non destinate ad essere impiegate come coloranti, e non impiegate conformemente alle condizioni indicate nel suddetto allegato.
d) conservanti:
i) conservanti diversi da quelli elencati nell’allegato V e conservanti ivi elencati ma non impiegati conformemente alle condizioni indicate nel suddetto allegato;
ii) fatte salve la lettera b, la lettera c), punto i) e la lettera e), punto i), sostanze elencate nell’allegato V ma non destinate ad essere impiegate come conservanti, e non impiegate conformemente alle condizioni indicate nel suddetto allegato.
e) filtri UV:
i) Filtri UV diversi da quelli elencati nell’Allegato VI e filtri UV ivi elencati ma non impiegati conformemente alle condizioni indicate nel suddetto allegato;
ii) fatte salve la lettera b), la lettera c), punto i), e la lettera d), punto i), sostanze elencate nell’allegato VI ma non destinate ad essere impiegate come filtri UV, e non impiegate conformemente alle condizioni indicate nel suddetto allegato.
2. In seguito ad una decisione della Commissione relativa all’estensione dell’ambito d’applicazione dell’allegato IV ai prodotti per la colorazione dei capelli, tali prodotti non possono contenere coloranti destinati a colorare i capelli diversi da quelli elencati nell’allegato IV e coloranti destinati a colorare i capelli che sono elencati in tale allegato ma non vengono impiegati in modo conforme alle condizioni ivi indicate.
La decisione della Commissione di cui al primo comma, volta a modificare elementi non essenziali del presente regolamento, è adottata secondo la procedura di regolamentazione con controllo di cui all’articolo 32, paragrafo 3.
In accordo all’allegato III, la presenza di idrochinone è vietata in tutti i prodotti cosmetici ad eccezione delle tinture per capelli e dei kit per unghie finte.

La Sicurezza dei recipienti PED non esposti a fiamma: la norma di riferimento EN 13445-1 2015
EN 13445-1:2015
Recipienti a pressione non esposti a fiamma - Parte 1: Generalità
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| EN_13445-1-2015 Annex A.pdf Annex A |
418 kB | 128 |

Ergonomia interfaccia uomo-macchina: I simboli
EN 61310-1
Sicurezza del macchinario.
Indicazione, marcatura e manovra.
Parte 1 - Prescrizioni per segnali visivi, acustici e tattili.
IEC 61310-1
Safety of machinery.
Indication, marking and actuation.
Part 1: requirements for visual, acoustic and tactile signals.
La Norma tratta le prescrizioni da applicarsi all’interfaccia uomo-macchina, per metodi visivi, acustici e tattili per indicare le informazioni relative alla sicurezza delle persone esposte.
Essa specifica i sistemi di colori, segnali, marcature ed altri avvisi previsti per indicare situazioni rischiose, codificando i segnali visivi, acustici e tattili di dispositivi indicatori e attuatori in modo da facilitare l’utilizzo sicuro dei macchinari.
La Norma è rivolta ai fornitori di macchine per le quali non esista alcuna norma di famiglia di prodotto o dedicata diprodotto.
La seconda edizione 2008 tiene conto dell’evoluzione della normativa internazionale.
Presunzione Conformità RESS Direttiva macchine 2006/42/CE
Allegato ZZB (informativo)
Requisiti Essenziali della Direttiva 2006/42/CE soddisfatti dalla Norma EN 61310-1.
La Norma Europea è stata preparata su mandato accordato al CENELEC dalla Commissione Europea e dall’Associazione Europea di Libero Scambio e, relativamente al suo campo di applicazione, soddisfa i seguenti requisiti essenziali tra quelli indicati nell’Allegato I della Direttiva 2006/42/CE:
- RESS 1.7.1
- RESS 1.7.1.1
- RESS 1.7.1.2
La conformità alla presente Norma costituisce un mezzo per soddisfare i requisiti essenziali della Direttiva/e interessata/e.
Il file raccoglie:
1. Focus Tecnico
2. Raccolta immagini jpg
3. File CEM importabile CM4 PRO
File CEM importabile in CEM4
Maggiori Info e acquisto Documento
Documento compreso nel Servizio di Abbonamento Marcatura CE
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| DT - EN 61310-1 Simboli ergonomia Interfaccia uomo-macchina - Estratto.pdf Simboli ergonomia Interfaccia uomo-macchina - Estratto |
578 kB | 603 | ||
| EN 61310-1 Simboli ergonomia Interfaccia uomo-macchina.zip Ergonomia Interfaccia uomo-macchina |
915 kB | 321 |
Report 30 del 29/07/2016 N.1 A12/0892/16 Germania
Approfondimento tecnico: Trucco permanente

Il prodotto “Dark Coffee”, marca “Golden Rose”, Mod. J01-01-13, è stato ritirato dal mercato perché non conforme alla Risoluzione del Consiglio d’Europa ResAP(2008)1 sui requisiti e criteri per la sicurezza dei tatuaggi e del trucco permanente.
Il prodotto contiene nickel (valore misurato 51.8 mg/kg). Il nichel può causare irritazione della pelle e/o suscitare reazioni allergiche. La Risoluzione del Consiglio d’Europa ResAP(2008)1 raccomanda che il contenuto di nichel, in inchiostri per tatuaggi ed in prodotti per il trucco permanente, sia il più basso possibile a livello tecnico.
Difatti, per quanto riguarda il grado di purezza delle preparazioni per tatuaggi e trucco permanente la normativa stabilisce i limiti di impurezza, indicati in tabella 3, ed adotta i limiti dei livelli consentiti di coloranti organici usati in generi alimentari e prodotti cosmetici stabiliti dalla Direttiva 95/45/EEC (La Direttiva 95/45/CE della Commissione, del 26 luglio 1995, stabilisce i requisiti di purezza specifici per le sostanze coloranti per uso alimentare.).
Tabella 3 Risoluzione del Consiglio d’Europa ResAP(2008)1
Concentrazioni di impurezza massime consentite nei prodotti per tatuaggi e per il trucco permanente (PMU)
|
Elemento o composto |
ppm |
ppb |
|
Arsenic (As) |
2 |
|
|
Barium (Ba) |
50 |
|
|
Cadmium (Cd) |
0.2 |
|
|
Cobalt (Co) |
25 |
|
|
Chromium (Cr) (VI) |
0.2 |
|
|
Copper (Cu) soluble |
25 |
|
|
Mercury (Hg) |
0.2 |
|
|
Nickel (Ni) |
As low as technically achievable |
|
|
Lead (Pb) |
2 |
|
|
Selenium (Se) |
2 |
|
|
Antimony (Sb) |
2 |
|
|
Tin (Sn) |
50 |
|
|
Zinc (Zn) |
50 |
|
|
Policyclic aromatic hydrocarbons (PAH) |
0.5 |
|
|
Benzene-a-pyrene (BaP) |
|
5 |
La presenza di tracce di nickel in prodotti per tatuaggi e PMU deve essere indicata sulla confezione insieme ad un’avvertenza (ad esempio, “Contiene nickel. Può causare reazioni allergiche.”).

ID 2844 | Update news 20.09.2023
Direttiva 2012/27/UE del Parlamento europeo e del Consiglio del 25 ottobre 2012 sull'efficienza energetica, che modifica le direttive 2009/125/CE e 2010/30/UE e abroga le direttive 2004/8/CE e 2006/32/CE.
(GU L 315/1 del 14.11.2012)
Attuata con: Decreto legislativo 4 luglio 2014, n. 102
[box-warning]Abrogazione dal 12.10.2025
Pubblicata in GU L 231/1 del 20.9.2023 la Direttiva (UE) 2023/1791 del Parlamento europeo e del Consiglio del 13 settembre 2023 sull'efficienza energetica e che modifica il regolamento (UE) 2023/955, con entrata in vigore il 10 ottobre 2023.
La Direttiva (UE) 2023/1791 abroga a decorrere dal 12 ottobre 2025 La direttiva 2012/27/UE.[/box-warning]
[box-warning]Update 24 Maggio 2023
Pubblicato in GU del 14 Aprile 2023 Il Regolamento delegato (UE) 2023/807 della Commissione del 15 dicembre 2022 che rivede il fattore di energia primaria per l’energia elettrica in applicazione della direttiva 2012/27/UE del Parlamento europeo e del Consiglio, in vigore il 04.05.2023[/box-warning]
[box-warning]Update 14 Giugno 2019
Pubblicata in GU del 14 Giugno 2019 la Direttiva (UE) 2019/944 del Parlamento Europeo e del Consiglio del 5 giugno 2019 relativa a norme comuni per il mercato interno dell'energia elettrica e che modifica la direttiva 2012/27/UE, entra in vigore il 04.07.2019[/box-warning]
[box-warning]Update 23 maggio 2019
Pubblicato in GU del 23 maggio 2019 il Regolamento delegato (UE) 2019/826 della Commissione, del 4 marzo 2019, che modifica gli allegati VIII e IX della direttiva 2012/27/UE del Parlamento europeo e del Consiglio sul contenuto delle valutazioni globali del potenziale di riscaldamento e raffreddamento efficienti, entra in vigore il 12 giugno 2019[/box-warning]
[box-warning]Update 19 giugno 2018
Pubblicata in GU del 19 giugno 2018 la Direttiva 2018/844 che modifica la direttiva 2010/31/UE sulla prestazione energetica nell’edilizia e la direttiva 2012/27/UE sull’efficienza energetica, in vigore dal 09 luglio p.v. e dovrà essere recepita dagli Stati membri entro il 10 marzo 2020.[/box-warning]
Attuata con: Decreto legislativo 4 luglio 2014, n. 102
________
Articolo 1 Oggetto e ambito di applicazione
1. La presente direttiva stabilisce un quadro comune di misure per la promozione dell'efficienza energetica nell'Unione al fine di garantire il conseguimento dell'obiettivo principale dell'Unione relativo all'efficienza energetica del 20 % entro il 2020 e di gettare le basi per ulteriori miglioramenti dell'efficienza energetica al di là di tale data.
Essa stabilisce norme atte a rimuovere gli ostacoli sul mercato dell'energia e a superare le carenze del mercato che frenano l'efficienza nella fornitura e nell'uso dell'energia e prevede la fissazione di obiettivi nazionali indicativi in materia di efficienza energetica per il 2020.
2. I requisiti stabiliti dalla presente direttiva sono requisiti minimi e non impediscono ai singoli Stati membri di mantenere o introdurre misure più rigorose. Tali misure sono compatibili con il diritto dell'Unione. Qualora la normativa nazionale preveda misure più rigorose, gli Stati membri notificano tale normativa alla Commissione.
...
segue
___________
Modificata da:
M1 - Direttiva 2013/12/UE del Consiglio del 13 maggio 2013 (GU L 141 28 28.5.2013)
M2 - Direttiva (UE) 2018/844 del Parlamento europeo e del Consiglio del 30 maggio 2018 (GU L 156 75 19.6.2018)
M3 - Direttiva (UE) 2018/2002 del Parlamento europeo e del Consiglio dell'11 dicembre 2018 (GU L 328 210 21.12.2018)
M4 - Regolamento (UE) 2018/1999 del Parlamento europeo e del Consiglio dell'11 dicembre 2018 (GU L 328 1 21.12.2018)
M5 - Decisione (UE) 2019/504 del Parlamento europeo e del Consiglio del 19 marzo 2019 (GU L 85I 66 27.3.2019)
M6 - Regolamento delegato (UE) 2019/826 della Commissione del 4 marzo 2019 (GU L 137 3 23.5.2019)
M7 - Direttiva (UE) 2019/944 del Parlamento europeo e del Consiglio del 5 giugno 2019 (GU L 158 125 14.6.2019) - Testo consolidato 01.01.2021
M8 - Regolamento delegato (UE) 2023/807 della Commissione del 15 dicembre 2022 (GU L 101/16 del 14.4.2023) - Testo consolidato 04.05.2023
Rettificata da:
C1 - Rettifica, GU L 113 del 25.4.2013, pag. 24 (2012/27/UE)
C2 - Rettifica, GU L 31 del 4.2.2020, pag. 10 (2018/2002)
C3 - Rettifica, GU L 387 del 19.11.2020, pag. 23 (2018/2002)
C4 - Rettifica, GU L 387 del 19.11.2020, pag. 24 (2018/2002)
Collegati:
[box-note]Esperti in Gestione dell’Energia e UNI CEI 11339
Decreto Legislativo 4 luglio 2014, n. 102
Direttiva 2009/125/CE
Regolamento delegato (UE) 2019/826
Efficienza energetica D.Lgs 4 luglio 2014 n. 102 | Consolidato[/box-note]

La Procedura più diffusa per gestire le operazioni di isolamento delle fonti di alimentazione in campo industriale conforme allo standard USA 29 CFR 1910.147.
Una procedura per l'isolamento delle fonti di energia largamente utilizzata in campo industriale è nota come Lockout/Tagout (LOTO); questa procedura è di origine statunitense ed è stata definita dall'Occupational Safety and Health Administration (OSHA) [www.osha.gov]
L’utilizzo della procedura di LockOut/TagOut (blocco del riavvio/denominazione: LOTO) sta prendendo piede a livello internazionale e non viene più considerata una soluzione di sicurezza da utilizzare solo negli Stati Uniti.
Il crescente numero di aziende a livello internazionale che implementano procedure LOTO indica chiaramente come l'industria tenda a seguire gli esempi di best practice.
Secondo la legislazione europea, l'implementazione di una procedura di LockOut/TagOut sul luogo di lavoro non è obbligatoria, tuttavia esistono speciali requisiti di legge che possono essere soddisfatti in maniera ottimale utilizzando una procedura LOTO.
Quando si implementa una procedura LOTO, questa deve essere conforme allo standard USA 29 CFR 1910.147 / ANSI/ASSE Z244.1
Questo standard prescrive che un'azienda rispetti i seguenti requisiti minimi:
1. Sviluppo di una strategia generale di LockOut/TagOut;
2. Definizione di un elenco di macchine che necessitano di una procedura di LockOut/TagOut;
3. Certezza della disponibilità dei dispositivi necessari per il blocco e il contrassegno di una macchina;
4. Sviluppo di una procedura di LockOut/TagOut per le macchine identificate;
5. Formazione del personale alla rilevazione di sorgenti di energia pericolose e all'implementazione della procedura LOTO.
In allegato Guide ed Esempi di Procedure LOTO
Collegati
[box-note]Procedura LOTO Disciplina | Format procedura
Procedura manutenzione LOTO (Lockout/Tagout): modello editabile con immagini
Procedura LOTO: Lockout/Tagout: isolamento fonti di alimentazione in campo industriale
Lockout / Tagout: ANSI/ASSE Z244.1 - 2016
ANSI ASSE Z244.1 Ed. 2016[/box-note]
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| LOTO Lockout Tagout Procedure.pdf Procedura per l\'isolamento delle fonti di energia |
16399 kB | 600 |

Edizione Aprile 2016
Nuova versione della 'Guida Blu', Aprile 2016, concernente l'applicazione delle Direttive di prodotto
E’ disponibile l'aggiornamento della Guida all'attuazione delle direttive fondate sul nuovo approccio e sull'approccio globale ("Guida Blu").
La nuova Guida Blu si basa sul contenuto della Guida per l'attuazione delle direttive fondate sul nuovo approccio e sull'approccio globale pubblicata nel 2000, ma riflette la modernizzazione che ha portato al quadro giuridico negli ultimi dieci anni.
Questa nuova versione della Guida include nuovi capitoli, per esempio, sugli obblighi degli operatori economici o di accreditamento, o capitoli completamente rivisti, come quelli in materia di normazione e di sorveglianza del mercato.
La nuova Guida Blu è stato redatta in stretta collaborazione con le autorità nazionali e le parti interessate.
Rev. 04.2014
Rev. 07.2015
Rev. 04.2016
[box-warning]29.06.2022 - Guida blu Ed. 2022
Pubblicata la Guida blu all'attuazione della normativa UE sui prodotti 2022
In allegato Guida blu 2022[/box-warning]
Articoli correlati
Le nuove Direttive UE 2014/2016
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| Guida blu attuazione della normativa UE sui prodotti 2022.pdf Ed. 2022 |
3512 kB | 17 | ||
| Guida Blu 2016.pdf Ed. 2016 |
1829 kB | 422 | ||
| Blue Guide 04.2016 EN.pdf Ed. 2016 |
2435 kB | 183 | ||
| Blue Guide 2014 Rev. 1.1 2015_EN.pdf Rev. 1.1 2015 |
2827 kB | 61 | ||
| Blue Guide 2014 EN.pdf Ed. 2014 |
3637 kB | 292 |
EN 280 Piattaforme di lavoro mobili elevabili - Ed. 2009
Calcoli per la progettazione - Criteri di stabilità - Costruzione - Sicurezza - Esami e prove
La norma specifica i requisiti tecnici e le misure di sicurezza per tutti i tipi e per tutte le dimensioni di piattaforme di lavoro elevabili, destinate a spostare persone alle posizioni di lavoro da cui possano svolgere mansioni dalla piattaforma di lavoro, con l'intendimento che le persone accedano ed escano dalla piattaforma di lavoro attraverso una posizione di accesso definita.
Requisiti:
4. Elenco dei pericoli
5. Requisiti e/o misure di sicurezza
6. Verifica dei requisiti e/o delle misure di sicurezza
7. Informazioni per l'uso
Attenzione: aggiornare con EN 280 Ed. 2013
UNI Edizione 2013: http://store.uni.com/magento-1.4.0.1/index.php/en-280-2013.html
Download file CEM sito cem4.eu
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| EN 280 2009 PLE Requisiti.pdf PLE |
132 kB | 159 |

Modello di Dichiarazione di Conformità UE RED
Rev. 2.0 2017
Direttiva 2014/53/UE - RED (Radio Equipment Directive)
Direttiva 2014/53/UE del Parlamento europeo e del Consiglio del 16 aprile 2014 concernente l'armonizzazione delle legislazioni degli Stati membri relative alla messa a disposizione sul mercato di apparecchiature radio e che abroga la direttiva 1999/5/CE
Articolo 1
Oggetto e ambito di applicazione
1. La presente direttiva istituisce un quadro normativo per la messa a disposizione sul mercato e la messa in servizio delle apparecchiature radio nell'Unione.
«apparecchiatura radio»: un prodotto elettrico o elettronico che emette e/o riceve intenzionalmente onde radio a fini di radiocomunicazione e/o radiodeterminazione o un prodotto elettrico o elettronico che deve essere completato con un accessorio, come un'antenna, per poter emettere e/o ricevere intenzionalmente onde radio a fini di radiocomunicazione e/o radiodeterminazione;
2. La presente direttiva non si applica alle apparecchiature elencate nell'allegato I:
1. Apparecchiature radio utilizzate da radioamatori ai sensi dell'articolo 1, definizione 56, delle norme radio dell'Unione internazionale delle telecomunicazioni (ITU), tranne nel caso in cui le apparecchiature siano state messe a disposizione sul mercato. Non sono considerati messi a disposizione sul mercato:
a) i kit di apparecchiature radio destinati a essere assemblati e utilizzati da radioamatori;
b) le apparecchiature radio modificate da radioamatori ad uso degli stessi;
c) le apparecchiature costruite da singoli radioamatori per scopi scientifici e sperimentali nel quadro dell'attività radioamatoriale.
2. Equipaggiamento marittimo che rientra nell'ambito di applicazione della direttiva 96/98/CE ( 1 ).
3. Prodotti per aerei, loro parti e pertinenze rientranti nell'ambito di applicazione dell'articolo 3 del regolamento (CE) n. 216/2008 del Parlamento europeo e del Consiglio ( 2 ).
4. Kit di valutazione su misura per professionisti, destinati a essere utilizzati unicamente in strutture di ricerca e sviluppo a tali fini.
3. La presente direttiva non si applica alle apparecchiature radio usate esclusivamente nelle attività concernenti la pubblica sicurezza, la difesa, la sicurezza dello Stato (compreso il benessere economico dello Stato, nel caso di attività riguardanti questioni connesse con la sicurezza dello Stato) e nelle attività dello Stato in materia di diritto penale. 4. Alle apparecchiature radio che rientrano nell'ambito di applicazione della presente direttiva non si applica la direttiva 2014/35/UE, fatta eccezione per l'articolo 3, paragrafo 1, lettera a), della presente direttiva.
ALLEGATO II
MODULO A DI VALUTAZIONE DELLA CONFORMITÀ CONTROLLO INTERNO DELLA PRODUZIONE
1. Il controllo interno della produzione è la procedura di valutazione della conformità con cui il fabbricante ottempera agli obblighi di cui ai punti 2, 3 e 4 del presente allegato e si accerta e dichiara, sotto la sua esclusiva responsabilità, che le apparecchiature radio interessate soddisfano i requisiti essenziali di cui all'articolo 3.
2. Documentazione tecnica Il fabbricante compila la documentazione tecnica conformemente all'articolo 21.
3. Produzione Il fabbricante prende tutte le misure necessarie affinché il processo di fabbricazione e il relativo controllo assicurino la conformità delle apparecchiature radio fabbricate alla documentazione tecnica di cui al punto 2 del presente allegato e ai requisiti essenziali pertinenti di cui all'articolo 3.
4. Marcatura CE e dichiarazione di conformità UE
4.1. Il fabbricante appone la marcatura CE a norma degli articoli 19 e 20 su ogni singola apparecchiatura radio conforme alle prescrizioni applicabili della presente direttiva.
4.2. Il fabbricante compila una dichiarazione scritta di conformità UE per ogni tipo di apparecchiatura radio che, insieme alla documentazione tecnica, tiene a disposizione delle autorità nazionali per dieci anni dalla data in cui l'apparecchiatura radio è stata immessa sul mercato. La dichiarazione di conformità UE identifica l'apparecchiatura radio per cui è stata compilata. Una copia della dichiarazione di conformità UE è messa a disposizione delle autorità competenti su richiesta.
5. Rappresentante autorizzato Gli obblighi del fabbricante di cui al punto 4 possono essere adempiuti dal suo rappresentante autorizzato, a nome del fabbricante e sotto la sua responsabilità, purché siano specificati nel mandato.
ALLEGATO III
MODULI B E C DI VALUTAZIONE DELLA CONFORMITÀ
ESAME UE DEL TIPO E CONFORMITÀ AL TIPO BASATA SUL CONTROLLO INTERNO DELLA PRODUZIONE
MODULO B Esame UE del tipo
1. L'esame UE del tipo è la parte di una procedura di valutazione della conformità con cui un organismo notificato esamina il progetto tecnico di un'apparecchiatura radio, nonché verifica e certifica che il progetto tecnico di tale apparecchiatura radio rispetta i requisiti essenziali di cui all'articolo 3.
2. L'esame UE del tipo è effettuato mediante la valutazione dell'adeguatezza del progetto tecnico dell'apparecchiatura radio effettuata esaminando la documentazione tecnica e la documentazione probatoria di cui al punto 3, senza esame di un campione (tipo di progetto).
3. Il fabbricante presenta una richiesta di esame UE del tipo a un unico organismo notificato di sua scelta. La domanda deve contenere:
a) il nome e l'indirizzo del fabbricante e, nel caso in cui la domanda sia presentata dal rappresentante autorizzato, il nome e l'indirizzo di quest'ultimo;
b) una dichiarazione scritta in cui si precisa che la stessa domanda non è stata presentata a nessun altro organismo notificato;
c) la documentazione tecnica che deve consentire di valutare la conformità dell'apparecchiatura radio alle prescrizioni applicabili della presente direttiva e comprende un'analisi e una valutazione adeguate dei rischi. La documentazione tecnica precisa le prescrizioni applicabili e include, se necessario ai fini della valutazione, il progetto, la fabbricazione e il funzionamento dell'apparecchiatura radio. Inoltre contiene, laddove applicabile, gli elementi di cui all'allegato V;
d) la documentazione probatoria attestante l'adeguatezza delle soluzioni del progetto tecnico. Tale documentazione cita tutti i documenti utilizzati, in particolare qualora le norme armonizzate pertinenti non sono state (integralmente) applicate, e comprende, se necessario, i risultati delle prove effettuate conformemente alle altre pertinenti specifiche tecniche dal laboratorio del fabbricante oppure da un altro laboratorio di prova, a nome e sotto la responsabilità del fabbricante.
4. L'organismo notificato esamina la documentazione tecnica e probatoria per valutare l'adeguatezza del progetto tecnico dell'apparecchiatura radio.
5. L'organismo notificato redige una relazione di valutazione che elenca le iniziative intraprese in conformità al punto 4 e i relativi risultati. Fatti salvi i suoi obblighi di cui al punto 8, l'organismo notificato rende pubblico l'intero contenuto della relazione, o parte di esso, solo con l'accordo del fabbricante.
6. Se il tipo risulta conforme alle prescrizioni della presente direttiva applicabili all'apparecchiatura radio in questione, l'organismo notificato rilascia al fabbricante un certificato di esame UE del tipo. Tale certificato riporta il nome e l'indirizzo del fabbricante, le conclusioni dell'esame, gli aspetti dei requisiti essenziali oggetto di esame, le eventuali condizioni di validità e i dati necessari per l'identificazione del tipo approvato. Il certificato di esame UE del tipo può comprendere uno o più allegati. Il certificato di esame UE del tipo e gli allegati devono contenere ogni utile informazione che permetta di valutare la conformità delle apparecchiature radio fabbricate al tipo esaminato e consentire il controllo del prodotto in funzione. Se il tipo non soddisfa i requisiti della presente direttiva a esso applicabili, l'organismo notificato rifiuta di rilasciare un certificato di esame UE del tipo e informa di tale decisione il richiedente, motivando dettagliatamente il suo rifiuto.
7. L'organismo notificato segue l'evoluzione del progresso tecnologico generalmente riconosciuto e valuta se il tipo approvato non è più conforme alle prescrizioni applicabili della presente direttiva. Esso decide se tale progresso richieda ulteriori indagini e in caso affermativo l'organismo notificato ne informa il fabbricante. Il fabbricante informa l'organismo notificato che detiene la documentazione tecnica relativa al certificato di esame UE del tipo di tutte le modifiche al tipo approvato, qualora possano influire sulla conformità dell'apparecchiatura radio ai requisiti essenziali della presente direttiva o sulle condizioni di validità di tale certificato. Tali modifiche comportano una nuova approvazione, sotto forma di un supplemento al certificato di esame UE del tipo.
8. Ogni organismo notificato informa la propria autorità di notifica dei certificati di esame UE del tipo e/o dei supplementi che esso ha rilasciato o revocato e, periodicamente o su richiesta, mette a disposizione dell'autorità di notifica l'elenco di tali certificati e/o dei supplementi respinti, sospesi o altrimenti sottoposti a restrizioni. Ogni organismo notificato informa gli altri organismi notificati dei certificati di esame UE del tipo e/o dei supplementi da esso respinti, ritirati, sospesi o altrimenti sottoposti a restrizioni, e, su richiesta, dei certificati e/o dei supplementi da esso rilasciati. Ogni organismo notificato informa gli Stati membri dei certificati di esame UE del tipo rilasciati e/o dei relativi supplementi nei casi in cui le norme armonizzate, i cui riferimenti sono stati pubblicati nella Gazzetta ufficiale dell'Unione europea, non siano state applicate (integralmente). La Commissione, gli Stati membri e gli altri organismi notificati possono ottenere, su richiesta, copia dei certificati di esame UE del tipo e/o dei relativi supplementi. La Commissione e gli Stati membri possono ottenere, su richiesta, copia della documentazione tecnica e dei risultati degli esami effettuati dall'organismo notificato. L'organismo notificato conserva una copia del certificato di esame UE del tipo, degli allegati e dei supplementi, nonché il fascicolo tecnico contenente la documentazione presentata dal fabbricante, per dieci anni a partire dalla valutazione dell'apparecchiatura radio o fino alla scadenza della validità di tale certificato.
9. Il fabbricante tiene a disposizione delle autorità nazionali una copia del certificato di esame UE del tipo, degli allegati e dei supplementi insieme alla documentazione tecnica per dieci anni dalla data in cui l'apparecchiatura radio è stata immessa sul mercato.
10. Il rappresentante autorizzato del fabbricante può presentare la richiesta di cui al punto 3 ed espletare gli obblighi di cui ai punti 7 e 9, purché siano specificati nel mandato.
MODULO C Conformità al tipo basata sul controllo interno della produzione
1. La conformità al tipo basata sul controllo interno della produzione fa parte di una procedura di valutazione della conformità con cui il fabbricante ottempera agli obblighi di cui ai punti 2 e 3, e si accerta e dichiara che le apparecchiature radio in questione sono conformi al tipo oggetto del certificato di esame UE del tipo e soddisfa i requisiti della presente direttiva a esso applicabili.
2. Fabbricazione Il fabbricante prende tutte le misure necessarie affinché il processo di fabbricazione e il suo controllo garantiscano la conformità delle apparecchiature radio prodotte al tipo oggetto del certificato di esame UE e ai requisiti applicabili della presente direttiva.
3. Marcatura CE e dichiarazione di conformità UE
3.1. Il fabbricante appone la marcatura CE a norma degli articoli 19 e 20 a ogni singola apparecchiatura radio conforme al tipo descritto nel certificato di esame UE del tipo e alle prescrizioni della presente direttiva ad essa applicabili.
3.2. Il fabbricante compila una dichiarazione scritta di conformità UE per ciascun tipo di apparecchiatura radio e la tiene a disposizione delle autorità nazionali per un periodo di dieci anni dalla data in cui l'apparecchiatura radio è stata immessa sul mercato. La dichiarazione di conformità UE identifica l'apparecchiatura radio per cui è stata compilata. Una copia della dichiarazione di conformità UE è messa a disposizione delle autorità competenti su richiesta.
4. Rappresentante autorizzato Gli obblighi del fabbricante di cui al punto 3 possono essere adempiuti dal suo rappresentante autorizzato, a nome del fabbricante e sotto la sua responsabilità, purché siano specificati nel mandato.
ALLEGATO IV
MODULO H DI VALUTAZIONE DELLA CONFORMITÀ CONFORMITÀ BASATA SULLA GARANZIA TOTALE DI QUALITÀ
1. La conformità basata sulla garanzia totale di qualità è la procedura di valutazione della conformità con cui il fabbricante che ottempera agli obblighi di cui ai punti 2 e 5, e si accerta e dichiara, sotto la sua esclusiva responsabilità, che l'apparecchiatura radio in questione risponde ai requisiti della presente direttiva a essa applicabili.
2. Produzione Il fabbricante applica un sistema approvato di qualità della progettazione, della fabbricazione, dell'ispezione delle apparecchiature radio finite e delle prove delle apparecchiature radio interessate, secondo quanto specificato al punto 3, ed è assoggettato alla sorveglianza di cui al punto 4.
3. Sistema di qualità
3.1. Il fabbricante presenta una domanda di verifica del suo sistema di qualità a un organismo notificato di sua scelta per le apparecchiature radio in questione. La domanda deve contenere:
a) il nome e l'indirizzo del fabbricante e, qualora la domanda sia presentata dal suo rappresentante autorizzato, il nome e l'indirizzo di quest'ultimo;
b) la documentazione tecnica per ciascun tipo di apparecchiatura radio che intende fabbricare. La documentazione tecnica contiene, laddove applicabile, gli elementi di cui all'allegato V;
c) la documentazione relativa al sistema di qualità; e
d) una dichiarazione scritta in cui si precisa che la stessa domanda non è stata presentata a nessun altro organismo notificato.
3.2. Il sistema di qualità deve garantire la conformità dell'apparecchiatura radio ai requisiti della presente direttiva a essa applicabili. Tutti i criteri, i requisiti e le disposizioni adottati dal fabbricante devono costituire una documentazione sistematica e ordinata sotto forma di misure, procedure e istruzioni scritte. Tale documentazione relativa al sistema di qualità deve consentire un'interpretazione uniforme di programmi, schemi, manuali e registri riguardanti la qualità. Essa deve includere in particolare un'adeguata descrizione:
a) degli obiettivi di qualità e della struttura organizzativa, delle responsabilità e dei poteri del personale direttivo in materia di progettazione e qualità del prodotto;
b) delle specifiche tecniche di progettazione, comprese le norme che saranno applicate e, qualora le relative norme armonizzate non siano applicate integralmente, dei mezzi per garantire che siano rispettati i requisiti essenziali della presente direttiva che si applicano alle apparecchiature radio;
c) delle tecniche di controllo e verifica della progettazione, dei processi e degli interventi sistematici per la progettazione delle apparecchiature radio rientranti nel tipo in questione; d) dei corrispondenti processi di fabbricazione, delle tecniche di controllo e di garanzia della qualità, dei processi e degli interventi sistematici che saranno applicati;
e) degli esami e delle prove che saranno effettuati prima, durante e dopo la fabbricazione, con indicazione della frequenza con cui si intende effettuarli;
f) dei registri riguardanti la qualità, come le relazioni ispettive e i dati sulle prove e sulle tarature, le relazioni sulle qualifiche del personale interessato ecc.;
g) dei mezzi di sorveglianza che consentono di controllare che sia ottenuta la qualità richiesta in materia di progettazione e di prodotti e se il sistema di qualità funziona efficacemente.
3.3. L'organismo notificato valuta il sistema di qualità per determinare se soddisfa i requisiti di cui al punto 3.2. Esso presume la conformità a tali requisiti degli elementi del sistema di qualità conformi alle specifiche corrispondenti delle pertinenti norme armonizzate. Oltre all'esperienza con i sistemi di gestione della qualità, almeno un membro del gruppo incaricato del controllo deve avere esperienza nella valutazione del settore e della tecnologia dell'apparecchiatura radio in questione e conoscere le prescrizioni applicabili della presente direttiva. Il controllo comprende una visita di valutazione dei locali del fabbricante. Il gruppo incaricato del controllo esamina la documentazione tecnica di cui al punto 3.1, lettera b), verifica la capacità del fabbricante di individuare le prescrizioni applicabili della presente direttiva e di effettuare gli esami atti a garantire la conformità delle apparecchiature radio a tali norme. La decisione è notificata al fabbricante o al suo rappresentante autorizzato. La notifica deve contenere le conclusioni del controllo e la motivazione circostanziata della decisione.
3.4. Il fabbricante deve impegnarsi a soddisfare gli obblighi derivanti dal sistema di qualità approvato e a fare in modo che esso rimanga adeguato ed efficace.
3.5. Il fabbricante deve tenere informato l'organismo notificato che ha approvato il sistema di qualità sulle modifiche che intende apportare al sistema di qualità. L'organismo notificato valuta le modifiche proposte e decide se il sistema modificato continui a soddisfare i requisiti di cui al punto 3.2 o se sia necessaria una nuova verifica. Esso notifica la decisione al fabbricante. La notifica deve contenere le conclusioni del controllo e la motivazione circostanziata della decisione.
4. Sorveglianza sotto la responsabilità dell'organismo notificato
4.1. Scopo della sorveglianza è garantire che il fabbricante soddisfi correttamente gli obblighi derivanti dal sistema di qualità approvato.
4.2. Il fabbricante deve consentire all'organismo notificato di accedere, ai fini della valutazione, ai locali di progettazione, fabbricazione, ispezione, prova e deposito fornendo tutte le necessarie informazioni, in particolare:
a) la documentazione relativa al sistema di qualità;
b) i registri riguardanti la qualità previsti dal sistema di qualità in materia di progettazione, come i risultati di analisi, calcoli, prove ecc.;
c) i registri riguardanti la qualità previsti dal sistema di qualità in materia di fabbricazione, come le relazioni ispettive e i dati sulle prove e sulle tarature, le relazioni sulle qualifiche del personale ecc.
4.3. L'organismo notificato deve svolgere controlli periodici intesi ad accertare che il fabbricante mantenga e applichi il sistema di qualità e fornisce al fabbricante una relazione sui controlli stessi.
4.4. Inoltre, l'organismo notificato può effettuare visite senza preavviso presso il fabbricante, procedendo o facendo procedere in tale occasione, se necessario, a prove sulle apparecchiature radio atte a verificare il corretto funzionamento del sistema di qualità. Esso deve fornire al fabbricante una relazione sulla visita e, se sono state svolte prove, una relazione sulle stesse.
5. Marcatura CE e dichiarazione di conformità UE
5.1. Il fabbricante appone la marcatura CE a norma degli articoli 19 e 20 e, sotto la responsabilità dell'organismo notificato di cui al punto3.1, il numero d'identificazione di quest'ultimo a ogni singola apparecchiatura radio conforme alle prescrizioni applicabili di cui all'articolo 3.
5.2. Il fabbricante compila una dichiarazione scritta di conformità UE per ciascun tipo di apparecchiatura radio e la tiene a disposizione delle autorità nazionali per dieci anni dalla data in cui l'apparecchiatura radio è stata immessa sul mercato. La dichiarazione di conformità UE identifica l'apparecchiatura radio per cui è stata compilata. Una copia della dichiarazione di conformità UE è messa a disposizione delle autorità competenti su richiesta.
6. Il fabbricante, per dieci anni a decorrere dalla data di immissione sul mercato dell'apparecchiatura radio, tiene a disposizione delle autorità nazionali:
a) la documentazione tecnica di cui al punto 3.1;
b) la documentazione relativa al sistema di qualità di cui al punto 3.1;
c) le modifiche di cui al punto 3.5 e la relativa approvazione;
d) le decisioni e le relazioni trasmesse dall'organismo notificato di cui ai punti 3.5, 4.3 e 4.4. 7.
Ogni organismo notificato informa la propria autorità di notifica delle approvazioni dei sistemi di qualità rilasciate o ritirate e, periodicamente o su richiesta, mette a sua disposizione l'elenco delle approvazioni dei sistemi di qualità respinte, sospese o altrimenti sottoposte a restrizioni. Ogni organismo notificato informa gli altri organismi notificati delle approvazioni dei sistemi di qualità da esso rifiutate, sospese o ritirate e, a richiesta, delle approvazioni del sistema di qualità rilasciate. 8. Rappresentante autorizzato Gli obblighi del fabbricante di cui ai punti 3.1, 3.5, 5 e 6 possono essere adempiuti dal suo rappresentante autorizzato, a nome del fabbricante e sotto la sua responsabilità, purché siano specificati nel mandato.
ALLEGATO VI
DICHIARAZIONE DI CONFORMITA' UE (N. XXXX)(*)
1. Modello di prodotto/prodotto (numero di prodotto, tipo, lotto o serie):
2. Nome e indirizzo del fabbricante e, se del caso, del suo rappresentante autorizzato:
3. La presente dichiarazione di conformità è rilasciata sotto la responsabilità esclusiva del fabbricante.
4. Oggetto della dichiarazione (identificazione del prodotto che ne consenta la tracciabilità; se necessario per l’identificazione del prodotto è possibile includere un’immagine):
5. L’oggetto della dichiarazione di cui sopra è conforme alla pertinente normativa di armonizzazione dell’Unione:
6. Riferimento alle pertinenti norme armonizzate utilizzate o riferimenti alle altre specifiche tecniche in relazione alle quali è dichiarata la conformità:
7. Se del caso, l’organismo notificato … (denominazione, numero) … ha effettuato (descrizione dell’intervento) … e rilasciato il certificato:
8. Informazioni aggiuntive: Firmato a nome e per conto di:
(luogo e data del rilascio): (nome, funzione) (firma):
(*) L’assegnazione di un numero, da parte del fabbricante, alla dichiarazione di conformità è opzionale.
ALLEGATO VII
DICHIARAZIONE DI CONFORMITÀ UE SEMPLIFICATA
La dichiarazione di conformità UE semplificata di cui all'articolo 10, paragrafo 9, deve essere presentata come segue: Il fabbricante, [nome del fabbricante], dichiara che il tipo di apparecchiatura radio [designazione del tipo di apparecchiatura radio] è conforme alla direttiva 2014/53/UE.
Il testo completo della dichiarazione di conformità UE è disponibile al seguente indirizzo Internet:
Elaborato Certifico S.r.l.
Rev. 1.0 2016 (Nuova Direttiva in vigore dal Luglio 2014 con applicazione definitiva il 20 Aprile 2016)
Rev. 2.0. 2017 (Inserimento campo immagine | foto prodotto)
Note
La Dichiarazione semplificata viene introdotta dal considerando 31 della Direttiva 2014/53/UE:
(31) L'obbligo stabilito dalla direttiva 1999/5/CE di includere una dichiarazione di conformità UE per le apparecchiature si è dimostrato utile ai fini della semplificazione e del miglioramento delle informazioni e dell'efficienza della vigilanza del mercato.
La possibilità di presentare una dichiarazione di conformità UE semplificata ha consentito una riduzione dell'onere collegato a tale obbligo senza diminuirne l'efficacia; è pertanto opportuno prevedere tale possibilità nella presente direttiva. Inoltre, al fine di garantire un accesso agevole ed efficiente alle dichiarazioni di conformità UE, ivi incluse quelle semplificate, dovrebbe essere possibile apporle sull'imballaggio dell'apparecchiatura radio interessata.
In base a quanto riportato nel considerando l’art. 10, par.9, stabilisce che:
I fabbricanti garantiscono che ogni singola apparecchiatura radio sia accompagnata da una copia della dichiarazione di conformità UE o da una dichiarazione di conformità UE semplificata. Se è fornita una dichiarazione di conformità UE semplificata, essa deve contenere l'esatto indirizzo Internet presso il quale è possibile ottenere il testo completo della dichiarazione di conformità UE.
Art. 18 DICHIARAZIONE DI CONFORMITÀ UE
[…]
La dichiarazione di conformità UE semplificata di cui all'articolo 10, paragrafo 9, contiene gli elementi di cui all'allegato VII ed è continuamente aggiornata. Essa è tradotta nella lingua o nelle lingue richieste dallo Stato membro nel quale l'apparecchiatura radio è immessa o messa a disposizione sul mercato. Il testo integrale della dichiarazione di conformità UE è disponibile sul sito Internet indicato nella dichiarazione di conformità UE semplificata, in una lingua o nelle lingue richieste dallo Stato membro nel quale l'apparecchiatura radio è immessa o messa a disposizione sul mercato.
[…]
Nuova Direttiva R&TTE 2014/53/UE (Direttiva RED)
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| Dichiarazione di conformità UE RED - Rev 2.0 2017.docx Certifico S.r.l. 2017 |
37 kB | 95 | ||
| Dichiarazione di conformità UE RED - Rev 0.0 2016.pdf Certifico S.r.l. 2016 |
325 kB | 63 | ||
| Dichiarazione di conformità UE RED - Rev 0.0 2016.docx Certifico S.r.l. 2016 |
35 kB | 31 |

Regolamento per l’attuazione della direttiva europea sugli ascensori e sui componenti di sicurezza degli stessi (Decreto del Presidente della Repubblica – esame preliminare)
1. Schema Decreto
2. Relazione illustrativa
3. Tabella di concordanza
Il Consiglio dei ministri, su proposta del Presidente Matteo Renzi, del Ministro dello sviluppo economico Carlo Calenda, del Ministro del lavoro e delle politiche sociali Giuliano Poletti e del Ministro per la semplificazione e la pubblica amministrazione Maria Anna Madia, ha deliberato l’approvazione in esame preliminare di un regolamento, da adottarsi con decreto del Presidente della Repubblica, concernente modifiche al decreto del Presidente della Repubblica del 30 aprile 1999, n. 162, per l’attuazione della direttiva 2014/33/UE relativa agli ascensori e ai componenti di sicurezza degli ascensori, nonché per l’esercizio degli ascensori.
Nello specifico, il provvedimento tiene conto delle innovazioni in materia di accreditamento degli organismi di valutazione della conformità, di vigilanza e controllo del mercato per quanto riguarda la commercializzazione dei prodotti, di principi generali della marcatura CE e di stato compatibile.
L’ambito di applicazione della direttiva si estende agli ascensori intesi come prodotti finiti e installati in modo permanente in edifici o costruzioni e ai componenti di sicurezza per ascensori nuovi prodotti da un fabbricante nell’Unione oppure componenti di sicurezza nuovi o usati importati da un paese terzo.
La direttiva 2014/33 responsabilizza gli operatori economici per la conformità degli ascensori e dei componenti di sicurezza per ascensori ai requisiti in essa previsti, in modo da garantire un elevato livello di protezione della salute e della sicurezza delle persone e dei beni, nonché una concorrenza leale sul mercato dell’Unione. Nell’interesse della competitività, è quindi fondamentale che gli organismi notificati applichino le procedure di valutazione della conformità senza creare oneri superflui per gli operatori economici.
Fonte: Comunicato stampa del Consiglio dei Ministri n. 121 20 Giugno 2016
Aggiornamento 15 Luglio 2016
Nuova Direttiva Ascensori: Direttiva 2014/33/UE
Il Decreto pubblicato: D.P.R. 10 gennaio 2017 n. 23
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| Schema Regolamento Ascensori 17 Giugno 2016.pdf 17 Giugno 2016 |
3194 kB | 65 |
Modifiche al Decreto Legislativo 4 marzo 2014, n. 27, recante attuazione della direttiva 2011/65/UE sulla restrizione dell’uso di determinate sostanze pericolose nelle apparecchiature elettriche ed elettroniche.
1. All’articolo 19 del decreto legislativo 4 marzo 2014, n. 27, sono apportate le seguenti modificazioni:
a) al comma 1, le parole: «e dal Ministero dell’ambiente e della tutela del territorio e del mare», sono sostituite dalle seguenti: «, dal Ministero dell’ambiente e
della tutela del territorio e del mare e dal Ministero della salute»;
b) al comma 1, dopo le parole: «e successive modificazioni» sono inserite le seguenti: «nelle more del riordino delle stesse ai sensi dell’articolo 10 della legge 7 agosto 2015, n. 124,»;
c) al comma 1, dopo le parole: «nonché dell’ISPRA» sono inserite le seguenti: «e dell’Istituto Superiore di Sanità (ISS).»;
d) dopo il comma 1 è aggiunto il seguente:
«1 -bis . I Ministeri dello sviluppo economico, dell’ambiente e della tutela del territorio e del mare e della salute svolgono le funzioni di cui al comma 1 sulla base di uno specifico protocollo d’intesa, in coordinamento con il “Comitato tecnico di Coordinamento” di cui all'articolo 7 del decreto del Ministro della salute del 22 novembre 2007, pubblicato nella Gazzetta Ufficiale n. 12 del 15 gennaio 2008, nonché in raccordo con le regioni e province autonome, ai fini del coordinamento tra le rispettive articolazioni organizzative, sulla base dei vigenti accordi in materia per gli ambiti di competenza.».
2. Il protocollo d’intesa di cui all’articolo 19, comma 1 -bis , del decreto legislativo 4 marzo 2014, n. 27, introdotto dal comma 1, lettera d) , del presente articolo, è sottoscritto entro sei mesi dalla data di entrata in vigore del presente decreto.
GU n.161 del 12.07.2016
Decreto Legislativo 4 marzo 2014, n. 27
Report 27 del 08/07/2016 N.1 A12/0816/16 Francia
Approfondimento tecnico: Etanolo per stufe senza canna fumaria/camino

Il prodotto, di marca DOMESTIX, è stato sottoposto alla procedura di ritiro dal mercato perché non conforme al Regolamento (CE) n . 1272/2008 del Parlamento europeo e del Consiglio del 16 dicembre 2008 relativo alla classificazione, all'etichettatura e all'imballaggio delle sostanze e delle miscele che modifica e abroga le direttive 67/548/CEE e 1999/45/CE e che reca modifica al regolamento (CE) n. 1907/2006 (CLP).
Il liquido può fuoriuscire quando la bottiglia viene capovolta a causa di un problema di fissaggio del tappo. Il prodotto è classificato come irritante per gli occhi e come infiammabile.
Il Regolamento (CE) n . 1272/2008 al Titolo IV “Imballaggio”, Articolo 35, stabilisce che:
1. Gli imballaggi contenenti sostanze o miscele pericolose sono soggetti alle seguenti prescrizioni:
a) l'imballaggio è concepito e realizzato in modo da impedire qualsiasi fuoriuscita del contenuto, tranne nei casi in cui sono prescritti speciali dispositivi di sicurezza;
b) i materiali che costituiscono l’imballaggio e la chiusura non debbono poter essere deteriorati dal contenuto, né poter formare con questo composti pericolosi;
c) tutte le parti dell’imballaggio e della chiusura sono solide e robuste, in modo da escludere qualsiasi allentamento e da sopportare in piena sicurezza le normali sollecitazioni di manipolazione;
d) gli imballaggi muniti di un sistema di chiusura che può essere riapplicato sono progettati in modo da poter essere richiusi varie volte senza fuoriuscite del contenuto.
Gli imballaggi contenenti una sostanza o miscela pericolosa fornita al pubblico non hanno una forma o un disegno che attiri o risvegli la curiosità attiva dei bambini o sia tale da indurre i consumatori in errore, né hanno una presentazione o un disegno simili a quelli utilizzati per prodotti alimentari, mangimi, medicinali o cosmetici, atti a indurre i consumatori in errore.
Se contiene una sostanza o miscela conforme alle disposizioni dell'allegato II, punto 3.1.1, l'imballaggio è munito di una chiusura di sicurezza per i bambini conforme alle disposizioni dell'allegato II, punti 3.1.2, 3.1.3 e 3.1.4.2.
Se contiene una sostanza o miscela conforme alle disposizioni dell'allegato II, punto 3.2.1, l'imballaggio è munito di un'indicazione di pericolo riconoscibile al tatto conforme alle disposizioni dell'allegato II, punto 3.2.2.
Se un detergente liquido per bucato destinato ai consumatori, quale definito all'articolo 2, paragrafo 1 bis, del regolamento (CE) n. 648/2004 del Parlamento europeo e del Consiglio (1), è contenuto in un imballaggio solubile monouso, si applicano i requisiti aggiuntivi di cui all'allegato II, punto 3.3.
3. L'imballaggio di sostanze e miscele è considerato conforme ai requisiti di cui al paragrafo 1, lettere a), b) e c), se soddisfa le norme in materia di trasporto di merci pericolose per via aerea, marittima, su strada, per ferrovia o per via fluviale.

Il presente rapporto tecnico internazionale ISO/TR 14121-2 (edizione giugno 2012), fornisce una guida pratica per l'esecuzione della valutazione del rischio per il macchinario in conformità alla UNI EN ISO 12100 e descrive diversi metodi e strumenti per ogni fase del processo.
Fornisce esempi di differenti misure che possono essere utilizzate per ridurre il rischio ed è destinata ad essere utilizzata per la valutazione del rischio di una estesa varietà di macchinari in termini di complessità e di potenziale di danno.
I suoi utilizzatori previsti sono le persone coinvolte nella progettazione, installazione o modifica del macchinario.
ISO/TR 14121-2 - Metodo Ibrido p.6.5
Download file CEM importabile in CEM4
Download file CEM importabile in CEM4 sito cem4.eu
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| ISO TR 14121-2-2012 - Estratto.pdf Estratto p. 6.5 Hybrid Tool |
308 kB | 674 |

ID 914 | Update news 31.12.2024 (Testo consolidato al 28.12.2024)
Direttiva 2014/53/UE del Parlamento europeo e del Consiglio del 16 aprile 2014 concernente l'armonizzazione delle legislazioni degli Stati membri relative alla messa a disposizione sul mercato di apparecchiature radio e che abroga la direttiva 1999/5/CE.
(GU L 153 del 22.5.2014)
[box-info]Recepita in data 14 Luglio 2016 con il D. Lgs. 128/2016
Decreto Legislativo 22 giugno 2016 n. 128
Attuazione della direttiva 2014/53/UE concernente l’armonizzazione delle legislazioni degli Stati membri relative alla messa a disposizione sul mercato di apparecchiature radio e che abroga la direttiva 1999/5/CE. (GU 163 del 14 Luglio 2016)[/box-info]
[box-warning]Update 11.09.2023
Pubblicato nella GU L 223/1 dell' 11.9.2023 il Regolamento delegato (UE) 2023/1717 della Commissione del 27 giugno 2023 che modifica la direttiva 2014/53/UE del Parlamento europeo e del Consiglio per quanto riguarda le specifiche tecniche per la presa di ricarica e per il protocollo di comunicazione per la ricarica per tutte le categorie o classi di apparecchiature radio che possono essere ricaricate mediante cavo
Entrata in vigore: 01.10.2023[/box-warning]
[box-warning]Update 07.12.2022
Pubblicata nella GU L 315/30 del 7.12.2022 la Direttiva (UE) 2022/2380 del Parlamento europeo e del Consiglio del 23 novembre 2022 che modifica la direttiva 2014/53/UE, concernente l’armonizzazione delle legislazioni degli Stati membri relative alla messa a disposizione sul mercato di apparecchiature radio
Entrata in vigore: 27.12.2022
Recepimento: entro il 28 dicembre 2023
Applicazione: a decorrere dal 28 dicembre 2024 per le categorie o classi di apparecchiature radio di cui all’allegato I bis, parte I, punti da 1.1. a 1.12., e dal 28 aprile 2026 per le categorie o classi di apparecchiature radio di cui all’allegato I bis, parte I, punto 1.13.[/box-warning]
[box-warning]Update 12.01.2022
Applicazione taluni Requisiti Essenziali direttiva RED vedi:
Regolamento delegato (UE) 2022/30 della Commissione del 29 ottobre 2021 che integra la direttiva 2014/53/UE del Parlamento europeo e del Consiglio per quanto riguarda l’applicazione dei requisiti essenziali di cui all’articolo 3, paragrafo 3, lettere d), e) ed f), di tale direttiva.(GU L 7/6 del 12.1.2022)[/box-warning]
La Direttiva RED (Radio Equipment Directive) 2014/53/UE, che HA sostituiTO la Direttiva R&TTE (Radio and Telecommunications Terminal Equipment) 1999/5/CE è applicata dal 13 giugno 2016 per regolamentare le apparecchiature radio al fine di apporre la Marcatura CE.
Date ufficiali e periodo di transizione
I prodotti Radio conformi ai requisiti essenziali di tale Direttiva potranno essere commercializzati liberamente all’interno dell’Unione Europea.
La direttiva RED è stata pubblicata sulla Gazzetta Ufficiale dell’Unione Europea il 22 maggio 2014.
Gli stati membri recepiranno la direttiva entro il 12 giugno 2016 con un periodo di transizione di un anno (13 Giugno 2017) per adeguarsi alla nuova direttiva.
Disposizioni transitorie
Per quanto riguarda gli aspetti contemplati dalla presente direttiva, gli Stati membri non ostacolano la messa a disposizione sul mercato o la messa in servizio delle apparecchiature radio oggetto della presente direttiva che sono conformi alla legislazione dell'Unione in materia applicabile anteriormente al 13 giugno 2016 e sono state immesse sul mercato anteriormente al 13 giugno 2017.
Recepimento
Gli Stati membri adottano e pubblicano, entro il 12 giugno 2016, le disposizioni legislative, regolamentari e amministrative necessarie per conformarsi alla presente direttiva.
Essi comunicano immediatamente alla Commissione il testo di tali disposizioni. Essi applicano tali disposizioni a decorrere dal 13 giugno 2016.
Abrogazione
La direttiva 1999/5/CE è abrogata a decorrere dal 13 giugno 2016.
![]()
Direttiva 2014/53/UE del Parlamento europeo e del Consiglio del 16 aprile 2014 concernente l'armonizzazione delle legislazioni degli Stati membri relative alla messa a disposizione sul mercato di apparecchiature radio e che abroga la direttiva 1999/5/CE (RED Radio Equipment Directive)
Direttiva 1999/5/CE del Parlamento europeo e del Consiglio, del 9 marzo 1999, riguardante le apparecchiature radio e le apparecchiature terminali di telecomunicazione e il reciproco riconoscimento della loro conformità (R&TTE Radio and Telecommunications Terminal Equipment)
[box-info]Recepita in data 14 Luglio 2016 con il D. Lgs. 128/2016
Decreto Legislativo 22 giugno 2016 n. 128
Attuazione della direttiva 2014/53/UE concernente l’armonizzazione delle legislazioni degli Stati membri relative alla messa a disposizione sul mercato di apparecchiature radio e che abroga la direttiva 1999/5/CE. (GU 163 del 14 Luglio 2016)[/box-info]
_______
Testo consolidato
Modifiche:
- Regolamento (UE) 2018/1139 del Parlamento europeo e del consiglio del 4 luglio 2018 - Consolidato 11.09.2018
- Direttiva (UE) 2022/2380 del Parlamento europeo e del Consiglio del 23 novembre 2022 - Consolidato 27.12.2022
- Regolamento delegato (UE) 2023/1717 della Commissione del 27 giugno 2023 - Consolidato 28.12.2024
- Direttiva (UE) 2024/2839 del Parlamento europeo e del Consiglio del 23 ottobre 2024 [...]
- Direttiva (UE) 2024/2749 del Parlamento europeo e del Consiglio del 9 ottobre 2024 (GU L 2024/2749 dell'8.11.2024) [...]
...
Collegati
[box-note]Regolamento delegato (UE) 2022/30
Direttiva R&TTE 1999/5/CE
Dichiarazione di Conformità UE Direttiva 2014/53/UE RED
Decreto 7 aprile 2017 n. 101
Decreto Legislativo 22 giugno 2016 n. 128[/box-note]

ID 1606 | Rev. 1.0 2019
Quando la valutazione dei rischi evidenzia che una macchina o un processo comporta un rischio di lesioni, il pericolo deve essere eliminato o contenuto.
Il modo in cui ottenere questo dipende dal tipo di macchina e di pericolo.
Le misure di protezione, in combinazione con una protezione fisica, possono prevenire l'accesso ad un pericolo o, quando l'accesso è possibile, il movimento pericoloso.
Esempi tipici di misure di protezione sono le protezioni fisiche interbloccate, le barriere fotoelettriche, le pedane di sicurezza, i controlli a due mani ed i dispositivi di abilitazione.
I dispositivi ed i sistemi di arresto di emergenza sono associati ai sistemi di controllo di sicurezza ma non sono sistemi di protezione diretta e dovrebbero essere considerati solo come misure di protezione complementari.
...
2. Prevenzione dell'accesso
2.1 Protezioni fisiche di chiusura Se il pericolo è su una parte della macchina a cui non bisogna accedere, è consigliabile installare sulla macchina una protezione permanente, come illustrato nella Figura 2.1.
Questi tipi di protezione, per essere rimossi, devono richiedere l'uso di attrezzi. Le protezioni fisse devono essere in grado di:
1) sopportare l'ambiente operativo,
2) contenere, quando necessario, i pezzi scagliati con violenza e
3) non creare pericoli presentando, per esempio, bordi taglienti. Le protezioni fisse possono avere aperture nei punti di attacco alla macchina o quando, per esempio, si tratta di recinzioni a griglia. Le finestre rappresentano un comodo modo di monitorare le prestazioni della macchina, quando si accede a quella parte della macchina. Bisogna, comunque, prestare attenzione alla scelta del materiale, dato che le interazioni chimiche con fluidi di taglio, raggi ultravioletti o il semplice invecchiamento possono provocare, nel tempo, il degrado del materiale della finestra. 
Figura 2.1: protezioni fisse
3. Dispositivi di rilevamento
Sono diversi i possibili dispositivi per rilevare la presenza di una persona che entra o si trova all'interno di una zona pericolosa. La scelta migliore per una particolare applicazione dipende da una serie di fattori.
- Frequenza di accesso,
- Tempo di arresto del pericolo,
- Importanza di fermare la macchina al completamento del ciclo, e
- Contenimento di pezzi scagliati violentemente, fluidi, nebbie, vapori, ecc.
Quando l'accesso al pericolo non è frequente, per proteggere contro pezzi scagliati con violenza, fluidi, nebbie ed altri tipi di pericoli, vengono spesso utilizzate protezioni mobili, adeguatamente selezionate ed interbloccate. Le protezioni interbloccate possono anche essere bloccate per impedire l'accesso alla macchina mentre il ciclo è ancora in corso e quando la macchina impiega molto tempo ad arrestarsi. I dispositivi di rilevamento della presenza, come barriere fotoelettriche, pedane e scanner, permettono un rapido e facile accesso alla zona pericolosa e vengono spesso scelti quando gli operatori devono accedere frequentemente alla zona di pericolo. Questi tipi di dispositivi non offrono protezione contro pezzi scagliati con violenza, nebbie, fluidi o altri tipi di pericoli. La misura di protezione migliore è un dispositivo o sistema che fornisca la massima protezione con il minimo intralcio al normale funzionamento della macchina. È necessario considerare tutti gli aspetti relativi all'utilizzo della macchina, dato che l'esperienza dimostra che un sistema difficile da usare è più soggetto ad essere rimosso o bypassato.
3.1 Dispositivi di rilevamento della presenza
Quando si decide come proteggere una zona o un'area, è importante capire esattamente quali funzioni di sicurezza sono richieste. In generale, ci saranno almeno due funzioni.
1. Spegnere o scollegare l'alimentazione quando una persona entra nella zona di pericolo.
2. Prevenire l'accensione o il ricollegamento dell'alimentazione quando una persona si trova all'interno della zona di pericolo. A prima vista, questi requisiti possono sembrare essere la stessa cosa ma, anche se sono correlati e spesso ottenuti con le stesse apparecchiature, in effetti si tratta di due funzioni separate. Per ottenere la prima funzione, dobbiamo usare qualche tipo di dispositivo di sgancio. In altre parole, un dispositivo che rileva il superamento di un certo punto, da parte di una persona, ed invia un segnale per sganciare l'alimentazione. Se la persona, successivamente, è in grado di proseguire oltre questo punto ma la sua presenza non viene più rilevata, è impossibile ottenere la seconda funzione (prevenzione dell'accensione).
...
La Figura 2.3 mostra un esempio di accesso completo del corpo con una barriera fotoelettrica montata verticalmente che funge da dispositivo di sgancio. Quando non c'è niente che ne impedisca la chiusura dopo l'entrata, anche le porte di protezione interbloccate possono essere considerate un dispositivo di solo sgancio.
Figura 2.3: accesso completo del corpo
...
Introduzione
Prevenzione dell'accesso
Dispositivi di rilevamento
Interruttori di sicurezza Interruttori con blocco della protezione
Interruttori di interblocco senza contatto
Interruttori a cerniera Interblocchi di posizione (interruttore di finecorsa)
Interblocchi a chiave bloccata
Dispositivi di interfaccia operatore
Dispositivi logici Controllori di sicurezza integrati
Reti di sicurezza
Dispositivi di uscita
Sistemi di collegamento
Fonte: Rockwell Automation
Pagine: 70
[box-note]Direttiva macchine 2006/42/CE
EN ISO 14119:2013 Interblocchi
ISO/TS 19837:2018 - Trapped Key Interlocking Devices [/box-note]
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| Macchine - Misure di protezione e dispositivi complementari - Rev. 1.0 2019.pdf Fonte: Rockwell Automation |
3497 kB | 268 | ||
| Macchine - Misure di protezione e dispositivi complementari - Rev. 0.0 2019.pdf Fonte: Rockwell Automation |
3721 kB | 402 |
Emendamenti del Parlamento europeo, approvati il 22 ottobre 2013, alla proposta di regolamento del Parlamento europeo e del Consiglio relativo ai dispositivi medici e recante modifica della direttiva 2001/83/CE, del regolamento (CE) n. 178/2002 e del regolamento (CE) n. 1223/2009 (COM(2012)0542 - C7-0318/2012 - 2012/0266(COD))

 2025/1234")

 Nuovi metodi di misurazione rumore dal 22.05.2025")




 2017/745 - (MDR)")




 305/2011 - CPR")
 2017/745")





