

![]()
|
|
![]()

Documenti
Legislazione
Direttive Sicurezza
Timeline Sicurezza IT
D.Lgs. 81/2008
Interpelli
Soggetti abilitati
ATECO / Livello rischio
Cassazione
Convenzioni ILO
TUSSL / Link


Documenti
Legislazione
Testo Unico Ambiente
Interpelli ambientali
Elenco CER
RENTRi


Documenti
Legislazione
CLP consolidato
REACH Consolidato
Seveso III
PXXX | EUH | HXXX
Elenco gas tossici
Parametri acque

Documenti
Legislazione
Norme armonizzate Click
Direttiva click
Nuovo approccio


Attività PI
D.P.R. 151/2011
Moduli PI
Codice RTO II
Norme PI 2022
Decreti PI Sett. 2021


Documenti
Legislazione
Tremcards
Etichette ADR
Kemler

Regolamento macchine
Direttiva macchine
Documenti Regolamento
Norme armonizzate
CEM4 | Software

HACCP (CE) 852/2004
MOCA (CE) 1935/2004
GMP (CE) 2023/2006
MOCA - GMP consolidato
Food Safety book

Documenti
Legislazione
DM. n. 37/2008
Legge n. 10/1991

Norme armonizzate
Direttiva click
Prassi di riferimento







| ID 4054 | | Visite: 261838 | Documenti Riservati Marcatura CE | Permalink: https://www.certifico.com/id/4054 |

ID 4054 | Rev. 1.2 del 15.03.2023 / Documento completo in allegato
Documento di sintesi, con schemi e tabelle, del nuovo Regolamento Dispositivi Medici (UE) 2017/745 (MDR), in pieno vigore dal 26 Maggio 2021.
Il nuovo Regolamento Dispositivi Medici (UE) 2017/745 (MDR), abroga la Direttiva 93/42/CEE (MDD) e s.m.i. entra in vigore il 25 Maggio 2017, con 1° step il 26.11.2017 (Organismi Notificati) e termine definitivo con abrogazione della direttiva 93/42/CEE (dispositivi medici) e direttiva 90/385/CEE (dispositivi medici impiantabili attivi), inserite entrambe nel regolamento, decorso dal 26 maggio 2021.
Vedi il testo consolidato del Regolamento MDR

Preview Focus - Nuovo Regolamento Dispositivi Medici (MDR) Rev. 1.2 2023
- Aggiunto capitolo 19 Specifiche comuni prodotti Allegato XVI Regolamento DM (Regolamento di esecuzione (UE) 2022/2346)
Regolamento di esecuzione (UE) 2022/2346 della Commissione del 1° dicembre 2022 che stabilisce specifiche comuni per i gruppi di prodotti che non hanno una destinazione d’uso medica elencati nell’allegato XVI.
Pubblicato in data 02.12.2022 il Regolamento di esecuzione (UE) 2022/2347 della Commissione del 1° dicembre 2022 recante modalità di applicazione del regolamento (UE) 2017/745 che pone deroga all’allegato VIII, punto 6.5, del regolamento (UE) 2017/745 per determinati gruppi di prodotti attivi che non hanno una destinazione d’uso medica elencati nell’allegato XVI.
- Aggiunto paragrafo 17.1 Monitoraggio e rivalutazione degli organismi notificati (art. 44 Regolamento DM modificato da Regolamento delegato (UE) 2023/502)
- Inserito riferimento/link Elenco Norme armonizzate Regolamento dispositivi medici 2017/745/CE
Regolamento (UE) 2020/561 del Parlamento Europeo e del Consiglio del 23 aprile 2020 che modifica il regolamento (UE) 2017/745 relativo ai dispositivi medici, per quanto riguarda le date di applicazione di alcune delle sue disposizioni.
Aggiunti paragrafi:
- Persona responsabile del rispetto della normativa
- Follow-up clinico post commercializzazione (PMCF)
- Registrazione degli operatori economici
- Organismi notificati
- Regole di classificazione e prodotti borderline
Novità rilevanti, rispetto alla precedente Direttiva, quali in sintesi:
- istituzione di una banca dati europea dispositivi medici (Eudamed)
- definizione e obblighi dettagliati di tutti gli operatori economici
- nuova figura del "Responsabile del rispetto della normativa"
- supervisione degli organismi notificati
- valutazione clinica, sorveglianza post-commercializzazione e piano PMCF (Post-Market Clinical Follow-up)
- trasparenza e la tracciabilità dei dispositivi medici (sistema UDI)
- dispositivi che contengono/costituiti da nanomateriali
Pubblicato in Gazzetta L 117/92 del 05 maggio 2017, il Regolamento (UE) 2017/745 del Parlamento e del Consiglio del 5 aprile 2017 relativo ai dispositivi medici, modifica:
- la direttiva 2001/83/CE (codice comunitario relativo ai medicinali per uso umano),
- il regolamento (CE) n. 178/2002 (sicurezza alimentare)
- il regolamento (CE) n. 1223/2009 (cosmetici)
e che abroga:
- la direttiva 90/385/CEE (dispositivi medici impiantabili attivi)
- la 93/42/CEE (dispositivi medici)
Vedi ebook MDR | Reg. (UE) 2017/745
Un atto giuridico unico per dispositivi medici impiantabili attivi e dispositivi medici (Art. 1)
Per ragioni storiche, i dispositivi medici impiantabili attivi, disciplinati dalla direttiva 90/385/CEE, e gli altri dispositivi medici, disciplinati dalla direttiva 93/42/CEE, erano disciplinati da due strumenti giuridici distinti. A fini di semplificazione entrambe le direttive, già modificate a più riprese, si sostituiscono con un unico atto legislativo, il Regolamento (UE) 2017/745, applicabile a tutti i dispositivi medici diversi dai dispositivi medico-diagnostici in vitro.
Persona responsabile del rispetto della normativa (Art. 15)
I fabbricanti, all'interno della loro organizzazione, dispongono di almeno una persona responsabile del rispetto della normativa che possieda le competenze necessarie nel settore dei dispositivi medici.
Le competenze necessarie sono attestate da una delle seguenti qualifiche:
a) un diploma, certificato o altro titolo ottenuto per aver completato studi universitari o un corso di studio riconosciuto equipollente dallo Stato membro in questione, in giurisprudenza, medicina, farmacia, ingegneria o un'altra disciplina scientifica pertinente, e almeno un anno di esperienza professionale nel campo della regolamentazione o dei sistemi di gestione della qualità relativi ai dispositivi medici;
b) quattro anni di esperienza professionale nel campo della regolamentazione o dei sistemi di gestione della qualità relativi ai dispositivi medici (per le microimprese tale figura può essere esterna).
Casi specifici (es medicinali e un dispositivo medico) (Art. 1)
I prodotti che combinano un medicinale o una sostanza e un dispositivo medico sono disciplinati dal presente regolamento o dalla direttiva 2001/83/CE del Parlamento europeo e del Consiglio (codice comunitario relativo ai medicinali per uso umano).
...
Obblighi di tutti gli operatori economici (Artt. 10, 11, 12, 13, 14, 16)
Sono definiti chiaramente gli obblighi generali dei diversi operatori economici:
- fabbricanti
- importatori
- distributori,
- operatore economico
- istituzione sanitaria
- utilizzatore
- utilizzatore profano
considerate anche le attività dei distributori come l'acquisizione, la detenzione e la fornitura di dispositivi.
Valutazione clinica e PMCF (Art. 61 e allegato XIV).
La valutazione clinica o le segnalazioni nell'ambito della vigilanza, che nelle direttive 90/385/CEE e 93/42/CEE erano definiti solo negli allegati, sono ora integrati nelle disposizioni del regolamento onde facilitarne l'attuazione.
I fabbricanti, quindi, devono effettuare una valutazione clinica ivi compreso un PMCF (Follow-up Clinico Post-Commercializzazione) (Art. 61 e allegato XIV).
Banca dati europea dispositivi medici: Eudamed (Art. 33 e 34)
Un aspetto fondamentale per il raggiungimento degli obiettivi del presente regolamento è la creazione di una Banca dati europea dei dispositivi medici (Eudamed) che integri diversi sistemi elettronici al fine di raccogliere ed elaborare le informazioni riguardanti i dispositivi presenti sul mercato e gli operatori economici, taluni aspetti della valutazione della conformità, gli organismi notificati, i certificati, le indagini cliniche, la vigilanza e la sorveglianza del mercato (Art. 33 e 34).
Gli obiettivi della banca dati sono: migliorare la trasparenza generale, anche grazie a un migliore accesso alle informazioni per il pubblico e gli operatori sanitari, evitare la moltiplicazione degli obblighi di informazione, rafforzare il coordinamento tra Stati membri e razionalizzare e facilitare il flusso di informazioni tra operatori economici, organismi notificati o sponsor e Stati membri, come pure tra gli stessi Stati membri e tra Stati membri e Commissione. Nel mercato interno questi obiettivi possono essere realizzati in maniera efficace solo al livello dell'Unione e la Commissione dovrebbe pertanto continuare a sviluppare e gestire la banca dati europea dei dispositivi medici istituita dalla decisione 2010/227/UE della Commissione.
Tracciabilità e sistema UDI (Art. 28)
La tracciabilità dei dispositivi grazie a un sistema di identificazione unica del dispositivo (sistema UDI), basato su linee guida internazionali, dovrebbe rafforzare considerevolmente l'efficacia delle attività legate alla sicurezza dopo la commercializzazione per i dispositivi, grazie a una migliore segnalazione degli incidenti, ad azioni correttive mirate di sicurezza e a una migliore sorveglianza da parte delle autorità competenti (Art. 28).
Direttiva EMC e Direttiva macchine (Art. 1)
Gli aspetti della sicurezza trattati dalla direttiva 2014/30/UE del Parlamento europeo e del Consiglio (EMC) sono parte integrante dei requisiti generali di sicurezza e prestazione stabiliti dal presente regolamento per i dispositivi. Il presente regolamento dovrebbe pertanto essere considerato una "lex specialis" rispetto a detta direttiva.
I dispositivi che sono anche macchine ai sensi dell'articolo 2, secondo comma, lettera a), della direttiva 2006/42/CE del Parlamento europeo e del Consiglio, laddove esista un rischio pertinente ai sensi di detta direttiva, rispettano altresì i requisiti essenziali in materia di salute e sicurezza stabiliti nell'allegato I di tale direttiva, qualora detti requisiti siano più specifici dei requisiti generali di sicurezza e prestazione stabiliti nell'allegato I, capo II, del presente regolamento (Art. 1).
Le Date (Artt. 122,123)
(*) Regolamento (UE) 2020/561: rinvio di un anno applicazione regolamento (UE) 2017/745
(**) Fatti salvi gli obblighi della Commissione ai sensi dell'articolo 34 (Funzionalità di Eudamed), qualora, a causa di circostanze che non avrebbero potuto essere ragionevolmente previste alla stesura del piano di cui all'articolo 34, paragrafo 1, Eudamed non sia pienamente operativa il 26 maggio 2021, gli obblighi e le prescrizioni relativi a Eudamed si applicano a decorrere dalla data corrispondente a sei mesi dalla data di pubblicazione dell'avviso di cui all'articolo 34, paragrafo 3 (pubblicazione GU avviso Eudamed).
...
Campo di applicazione (Art. 1)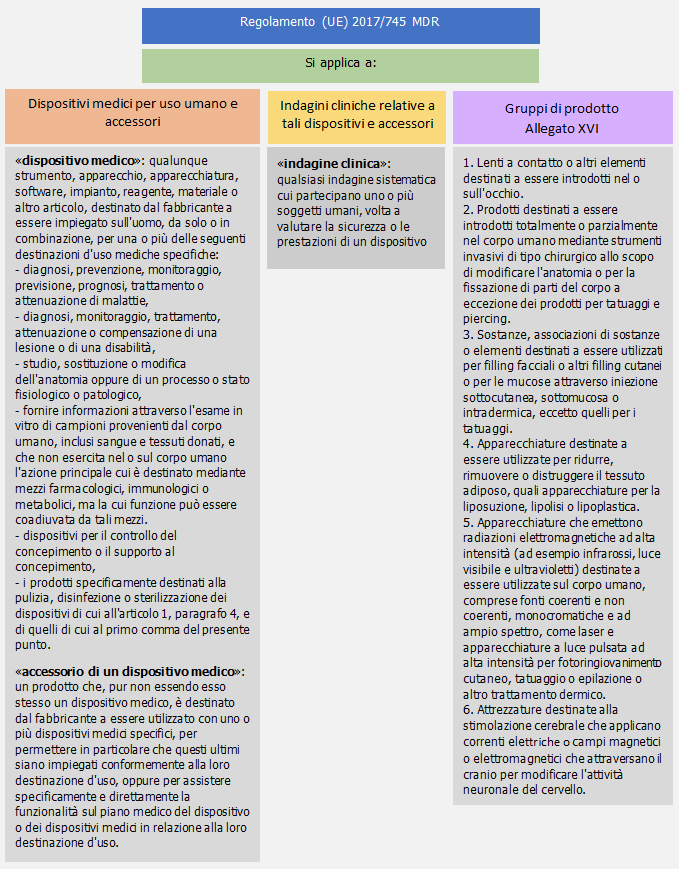
...
Classificazione (All. VIII)
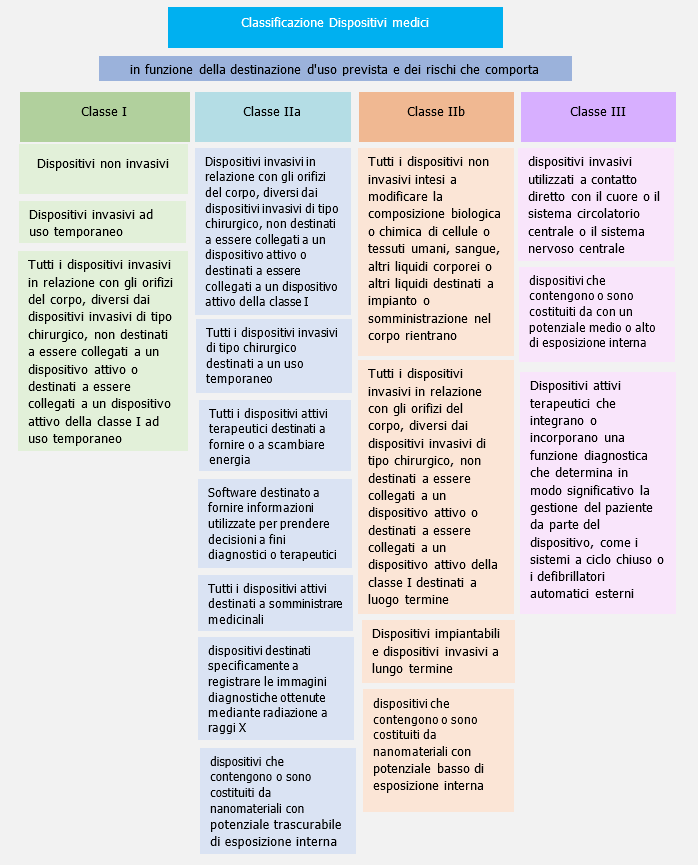
Segue
Indice
1. Premessa
2. Entrata in vigore e applicazione (Rev. 1.1 2020)
3. Oggetto e ambito di applicazione
4. Classificazione dei dispositivi
5. Obblighi attori MDR
5.1 Obblighi dei fabbricanti
5.2 Obblighi mandatario
5.3 Obblighi generali degli importatori
5.4 Obblighi generali dei distributori
5.5 Casi in cui gli obblighi dei fabbricanti si applicano agli importatori, ai distributori o ad altre persone
6. Persona responsabile del rispetto della normativa” (PR o PRRC da Person Responsible for Regulatory Compliance) (Rev. 1.0 2020)
7. Immissione sul mercato e messa in servizio
8. Requisiti generali di sicurezza e prestazione Allegato I
8.1 Requisiti generali
8.2 Requisiti relativi alla progettazione e alla fabbricazione
8.2.1 Caratteristiche chimiche, fisiche e biologiche
8.2.2 Infezione e contaminazione microbica
8.2.3 Dispositivi contenenti una sostanza considerata un prodotto medicinale e dispositivi che sono costituiti da sostanze o da una combinazione di sostanze che sono assorbite dal corpo umano o in esso localmente disperse
8.2.4 Dispositivi contenenti materiali di origine biologica
8.2.5 Fabbricazione dei dispositivi e interazione con il loro ambiente
8.2.6 Dispositivi con funzione diagnostica o di misura
8.2.7 Protezione contro le radiazioni
8.2.8 Sistemi elettronici programmabili - dispositivi contenenti sistemi elettronici programmabili e software che costituiscono dispositivi a sé stanti
8.2.9 Dispositivi attivi e dispositivi a essi collegati
9. Ricorso a norme armonizzate (Rev. 1.2 2023)
10. Specifiche comuni
11. Procedure di valutazione della conformità
11.1 Allegato IX Valutazione della conformità basata sul sistema di gestione della qualità e sulla valutazione della documentazione tecnica
11.2 Allegato X Valutazione della conformità basata sull'esame di tipo
11.3 Allegato XI Valutazione della conformità basata sull'assicurazione di qualità della produzione
12. Valutazione clinica
13. Follow-up clinico post commercializzazione (PMCF) (Rev. 1.0 2020)
14. Dichiarazione di conformità UE Allegato IV
15. Marcatura CE di conformità Allegato V
16. Registrazione degli operatori economici (Rev. 1.0 2020)
17. Organismi notificati (Rev. 1.0 2020)
17.1 Monitoraggio e rivalutazione degli organismi notificati (Rev. 1.2 2023)
18. Regole di classificazione e prodotti borderline (Rev. 1.0 2020)
19. Specifiche comuni prodotti Allegato XVI Regolamento DM (Rev. 1.2 2023)
...
Vedi il testo consolidato del Regolamento MDR
Certifico Srl - IT Rev. 1.2. 2023
Copia autorizzata Abbonati
Matrice Revisioni
| Rev. | Data | Oggetto | Autore |
| 1.2 | 15.03.2023 | - Aggiunto paragrafo 19 Specifiche comuni prodotti Allegato XVI Regolamento DM (Regolamento di esecuzione (UE) 2022/2346) - Aggiunto paragrafo 17.1 Monitoraggio e rivalutazione degli organismi notificati (art. 44 Regolamento DM modificato da Regolamento delegato (UE) 2023/502) - Inserito riferimento/link Elenco Norme armonizzate MDR 2017/745/CE |
Certifico Srl |
| 1.1 | 24.04.2020 | Regolamento (UE) 2020/561 | Certifico Srl |
| 1.0 | 20.03.2020 | Aggiunti paragrafi: - Persona responsabile del rispetto della normativa - Follow-up clinico post commercializzazione (PMCF) - Registrazione degli operatori economici - Organismi notificati - Regole di classificazione e prodotti borderline |
Certifico Srl |
| 0.0 | 23.05.2017 | Certifico Srl |
Collegati:
| Descrizione | Livello | Dimensione | Downloads | |
|---|---|---|---|---|
| Focus - Nuovo Regolamento Dispositivi Medici (MDR) Rev. 1.2 2023.pdf Certifico Srl - Rev. 1.2 2023 |
764 kB | 60 | ||
| Focus - Nuovo Regolamento Dispositivi Medici (MDR) Rev. 1.1 2020.pdf Certifico Srl - Rev. 1.1 2020 |
1069 kB | 246 | ||
| Focus - Nuovo Regolamento Dispositivi Medici (MDR) Rev. 1.0 2020.pdf Certifico Srl - Rev. 1.0 2020 |
1063 kB | 154 | ||
| Il Nuovo Regolamento Dispositivi Medici Rev. 00 2017- MDR.pdf Certifico Srl - Rev. 0.0 2017 |
630 kB | 284 |
| Descrizione | Lingua | Dimensione | Downloads | |
|---|---|---|---|---|
| Preview Focus - Nuovo Regolamento Dispositivi Medici (MDR) Rev. 1.2 2023 Certifico Srl - Rev. 1.2 2023 | IT | 426 kB | 1390 | |
| Preview Focus Regolamento Dispositivi Medici (MDR) Rev. 1.1 2020 Certifico Srl - Rev. 1.1 2020 | IT | 496 kB | 6540 | |
| Preview Focus - Nuovo Regolamento Dispositivi Medici (MDR) Rev. 1.0 2020 Certifico Srl - Rev. 1.0 2020 | IT | 500 kB | 3215 | |
| Regolamento Dispositivi Medici Rev. 00 2017- MDR Campo applicazione Certifico Srl. - Rev. 0.0 2017 | IT | 210 kB | 6631 |
Tags: Marcatura CE Abbonati Marcatura CE Regolamento Dispositivi Medici




















