|

EN ISO 14971 Dispositivi medici: Valutazione del rischio
Il 25 Maggio 2017 sono entrati in vigore i nuovi Regolamenti sui Dispositivi medici Regolamento (UE) 2017/745 (MDR) e Regolamento (UE) 2017/746 (IVDR) che si sapplicano con decorrenza principale rispettivamente dal 26 Maggio 2020 e 26 Maggio 2022.
La EN ISO 14971:2012, armonizzata IVD e MD, norma base per gestione dei rischi dei dispositivi medici, ha già in effetti anticipato determinati aspetti procedurali di gestione del rischio consoni (Presunzione di Conformità) ai nuovi Regolamenti.
Vedi il Documento sul Regolamento MDR:
Il Regolamento (UE) 2017/745 (MDR)
Il Documento è estratto dalla norma EN ISO 14971:2012 (UNI CEI EN ISO 14971:2012) Dispositivi medici - Applicazione della gestione dei rischi ai dispositivi medici, che è armonizzata per:
1. la Direttiva 93/42/CEE Dispositivi Medici sostituita dal Regolamento (UE) 2017/745 (MDR)
2. la Direttiva 90/385/CEE Dispositivi Medici Impiantabili Attivi sostituita dal Regolamento (UE) 2017/745 (MDR)
3. la Direttiva 98/79/CE Dispositivi Medico-Diagnostici in Vitro (IVD) sostituita dal Regolamento (UE) 2017/746 (IVDR)
La EN ISO 14971, insieme alle:
- IEC 60601-1 (sicurezza elettrica),
- ISO 13485 (Sistemi Gestione Qualità),
- IEC/EN 62366 (Usabilità dei dispositivi medici),
- ISO 10993 (valutazione biologica)
- IEC 62304 (software dispositivo medico).
definiscono la base degli standard per la sicurezza dei dispositivi medici.
Il Documento, estratto EN ISO 14971, intende fornire gli elementi essenziali per la Procedura di Valutazione dei rischi per i Dispositivi Medici, nel termine più ampio di "Gestione dei rischi" (Vedi Figura B.1 Schema delle attività di gestione del rischio come applicato ai dispositivi medici), fornendo le informazioni essenziali con schemi ed esempi di tabelle di stima del rischio.
I punti trattati nel documento sono inerenti ai punti della norma:
4. ANALISI DEL RISCHIO
4.1 Uso previsto
4.2 Identificazione dei pericoli
4.3 Stima dei rischi
5. VALUTAZIONE DEL RISCHIO
6 CONTROLLO DEL RISCHIO
6.1 Riduzione del rischio
6.2 Analisi delle opzioni di controllo del rischio
6.3 Implementazione delle misure di controllo del rischio
6.4 Valutazione del rischio residuo
6.5 Analisi rischi/benefici
6.6 Rischi derivanti dalle misure di controllo del rischio
6.7 Completezza del controllo del rischio
7. VALUTAZIONE DELL'ACCETTABILITÀ DEL RISCHIO RESIDUO COMPLESSIVO
8. RAPPORTO DI GESTIONE DEL RISCHIO
9. INFORMAZIONI DI PRODUZIONE E POST-PRODUZIONE
APPENDICE B (informativa)
PANORAMICA DEL PROCESSO DI GESTIONE DEL RISCHIO PER I DISPOSITIVI MEDICI
APPENDICE C (informativa)
QUESITI UTILIZZABILI PER IDENTIFICARE LE CARATTERISTICHE DEL DISPOSITIVO MEDICO CHE POTREBBERO AVERE UN IMPATTO SULLA SICUREZZA
APPENDICE D (informativa)
CONCETTI DI RISCHIO APPLICATI Al DISPOSITIVI MEDICI
La norma specifica una procedura che permette al fabbricante di identificare i pericoli associati ai dispositivi medici, inclusi i dispositivi medico-diagnostici in vitro, per stimare e valutare i rischi associati, per controllare tali rischi, e per monitorare l'efficacia dei controlli.
I requisiti della norma sono applicabili a tutte le fasi del ciclo di vita di un dispositivo medico.
La norma non si applica al processo decisionale clinico e non specifica i livelli di rischio accettabili.
Excursus
...
3.5 Documentazione di gestione del rischio
Per il particolare dispositivo medico preso in considerazione, il fabbricante deve definire e mantenere una documentazione di gestione del rischio. Oltre ai requisiti degli altri punti della presente norma internazionale, la documentazione di gestione del rischio deve fornire la rintracciabilità per ogni pericolo identificato per:
- l'analisi dei rischi;
- la valutazione dei rischi;
- l'attuazione e la verifica delle misure di controllo dei rischi;
- l’accertamento dell'accettabilità degli eventuali rischi residui.
4. ANALISI DEL RISCHIO
Deve essere eseguita l'analisi del rischio per il particolare dispositivo medico come descritto nei punti da 4.2 a 4.4. La conduzione e i risultati delle attività di analisi pianificata del rischio devono essere registrati nella documentazione di gestione del rischio.
...
5. VALUTAZIONE DEL RISCHIO
Per ogni situazione pericolosa identificata, il fabbricante deve decidere, impiegando i criteri definiti nel piano di gestione del rischio, se è necessario perseguire la riduzione del rischio. Se la riduzione del rischio non è richiesta, i requisiti forniti dal punto 6.2 al punto 6.6 non si applicano per questa situazione pericolosa (ovvero passare al punto 6.7). I risultati di questa valutazione del rischio devono essere registrati nella documentazione di gestione del rischio.
....
6. CONTROLLO DEL RISCHIO
...
Il fabbricante deve identificare la(e) misura(e) di controllo del rischio appropriata(e) per ridurre il(i) rischio(i) a un livello accettabile.
Il fabbricante deve utilizzare uno o più degli elementi seguenti di controllo del rischio nell'ordine di priorità elencato:
a) sicurezza inerente mediante progettazione;
b)misure protettive nel dispositivo medico stesso o nel processo di fabbricazione;
c) informazioni per la sicurezza.
...
7 VALUTAZIONE DELL'ACCETTABILITÀ DEL RISCHIO RESIDUO COMPLESSIVO
Quando sono state attuate e verificate tutte le misure di controllo del rischio, il fabbricante deve decidere se il rischio residuo complessivo posto dal dispositivo medico è accettabile impiegando i criteri definiti nel piano di gestione del rischio.
...
8. RAPPORTO DI GESTIONE DEL RISCHIO
Prima del rilascio per la distribuzione commerciale del dispositivo medico, il fabbricante deve eseguire un riesame del processo di gestione del rischio. Questo riesame deve assicurare come minimo che:
- il piano di gestione del rischio è stato adeguatamente attuato;
- il rischio residuo complessivo è accettabile;
- siano in atto metodi appropriati al fine di ottenere informazioni pertinenti di produzione e post-produzione.
9 INFORMAZIONI DI PRODUZIONE E POST-PRODUZIONE
Il fabbricante deve stabilire, documentare e mantenere una procedura sistematica per raccogliere e riesaminare le informazioni ottenute sul dispositivo medico o dispositivi simili nelle fasi di produzione e post-produzione.
...
APPENDICE B (informativa)
PANORAMICA DEL PROCESSO DI GESTIONE DEL RISCHIO PER I DISPOSITIVI MEDICI
La figura B.1, a carattere illustrativo, fornisce una panoramica del processo di gestione del rischio.
Come indicato, il processo deve essere iterativo, coprire ciascuno dei rischi e riportare alle fasi precedenti qualora le misure di controllo del rischio introducano nuovi pericoli o qualora divengano disponibili nuove informazioni.
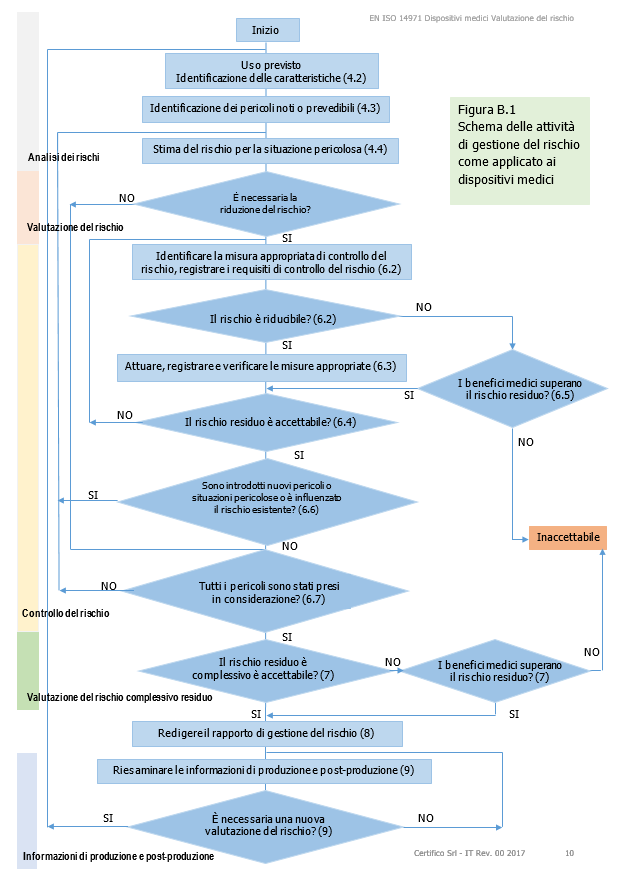 Figura B.1 Schema delle attività di gestione del rischio come applicato ai dispositivi medici Figura B.1 Schema delle attività di gestione del rischio come applicato ai dispositivi medici
...
APPENDICE C (informativa)
QUESITI UTILIZZABILI PER IDENTIFICARE LE CARATTERISTICHE DEL DISPOSITIVO MEDICO CHE POTREBBERO AVERE UN IMPATTO SULLA SICUREZZA
C.1 Generalità
Il sottopunto 4.2 richiede che il fabbricante identifichi quelle caratteristiche del dispositivo medico che potrebbero avere un impatto sulla sicurezza. La considerazione di queste caratteristiche è una fase essenziale per l'identificazione dei pericoli del dispositivo medico come richiesto nel punto 4.3. Un modo è quello di porre una serie di domande riguardanti la fabbricazione, gli utilizzatori previsti, l'uso previsto, l'uso improprio ragionevolmente prevedibile e lo smaltimento finale del dispositivo medico. Se si pongono tali domande dal punto di vista di tutti gli individui coinvolti (ad esempio utilizzatori, addetti alla manutenzione, pazienti, ecc.), può emergere un quadro più completo di dove siano reperibili i potenziali pericoli.
APPENDICE D (informativa)
CONCETTI DI RISCHIO APPLICATI Al DISPOSITIVI MEDICI
...
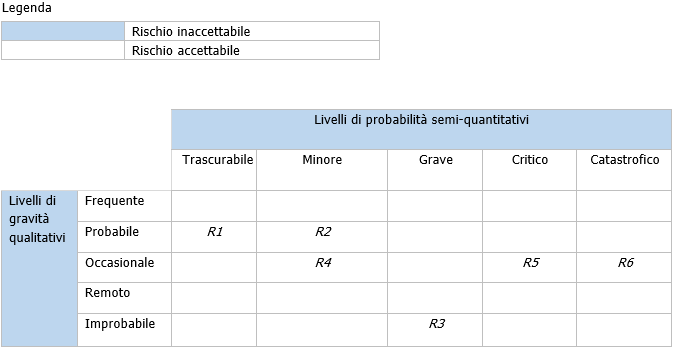
Figura D.5 Esempio di una matrice di valutazione del rischio semi-quantitativa
...
D5. Controllo del rischio
...
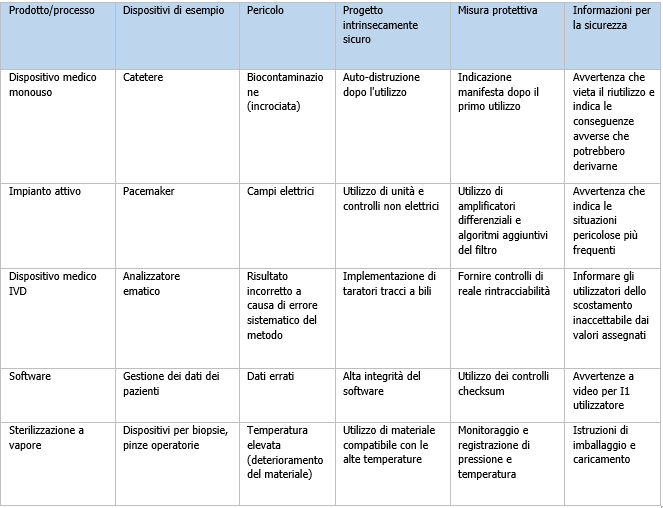
Figura D.6 Alcuni esempi di misure di controllo del rischio
...
segue
Fonte:
UNI CEI EN ISO 14971:2012
Dispositivi medici - Applicazione della gestione dei rischi ai dispositivi medici
Pag. 26
Riservato Abbonati: Normazione, 3X, 4X, Full
Certifico Srl - IT Rev. 00 2017
Info e download
|