

~ 2000 / 2026 ~
// Documenti disponibili n:
48.702
// Documenti scaricati n:
40.913.022
// Newsletter n:
3806


















Dichiarazione Art. 22 MDR Sistemi e Kit procedurali / Note e Modello
ID 3641
Per poter visualizzare la newsletter con i dati personalizzati devi essere connesso. Fai click qui per effettuare l'accesso al sito.
| Appunti Marcatura CE | ||
| Newsletter n. 3574 del 13 Luglio 2026 | ||
| Salve Visitatore | ||
Dichiarazione Art. 22 MDR Sistemi e Kit procedurali / Note e Modello ID 26669 | 12 Luglio 2026 / Allegato La dichiarazione Art. 22 del Regolamento (UE) 2017/745 (MDR) è il documento ufficiale che permette a chi assembla sistemi o kit procedurali (es. set chirurgici) di immetterli sul mercato europeo senza una nuova marcatura CE, assumendosi la responsabilità della loro compatibilità e sicurezza. Non esiste un modello ufficiale, tuttavia, la dichiarazione deve essere redatta per iscritto e contenere talune informazioni minime (vedi a seguire). L'Assemblatore di Sistemi e kit procedurali (System/Procedure Pack Producer), dovrà considerare che: - E' possibile combinare solo dispositivi già marcati CE / DMDV e altri prodotti compatibili (Art. 22 co. 1 lett. c)), seguendo le istruzioni dei rispettivi fabbricanti. - Se il kit include dispositivi da sterilizzare prima dell'uso, l'assemblatore deve applicare specifici procedure (Allegato IX o XI, parte A dell'MDR) e dichiararlo. - Il kit non ha una marcatura CE propria. Deve riportare però i dati dell'assemblatore, che deve fornire tutte le istruzioni originali ai pazienti/operatori. Modello della Dichiarazione (Fac-simile) con informazioni minime:
[...] Articolo 2 Definizioni 11) «sistema»: una combinazione di prodotti, confezionati insieme o non, che sono destinati a essere interconnessi o combinati per raggiungere una specifica destinazione d'uso medica; Art. 22 Sistemi e kit procedurali 1. Ogni persona fisica o giuridica deve redigere una dichiarazione che combina dispositivi recanti la marcatura CE con i seguenti altri dispositivi o prodotti, in maniera compatibile con la destinazione d'uso dei dispositivi o degli altri prodotti e nei limiti di utilizzo previsti dai loro fabbricanti, per immetterli sul mercato come sistema o kit procedurale: a) altri dispositivi recanti la marcatura CE; b) dispositivi medico-diagnostici in vitro recanti la marcatura CE conformemente al regolamento (UE) 2017/746; c) altri prodotti conformi alla normativa a essi applicabile solo qualora siano utilizzati nell'ambito di una procedura medica o ne sia altrimenti giustificata la presenza nel sistema o kit procedurale. 2. Nella dichiarazione a norma del paragrafo 1, la persona fisica o giuridica in questione indica: a) di aver verificato la compatibilità reciproca dei dispositivi e, se del caso, degli altri prodotti secondo le istruzioni dei fabbricanti e di aver effettuato le sue attività secondo tali istruzioni; b) di aver imballato il sistema o kit procedurale e fornito agli utilizzatori le relative informazioni, comprese le informazioni che devono essere fornite dai fabbricanti dei dispositivi o degli altri prodotti che sono stati assemblati; c) che l'attività di combinare dispositivi e, se del caso, altri prodotti come sistemi o kit procedurali è stata sottoposta a metodi adeguati di controllo interno, verifica e convalida. 3. Ogni persona fisica o giuridica che, ai fini della loro immissione sul mercato, sterilizza i sistemi o kit procedurali di cui al paragrafo 1 applica, a sua scelta, una delle procedure di cui all'allegato IX o la procedura di cui all'allegato XI, parte A. L'applicazione di tali procedure e l'intervento dell'organismo notificato si limitano agli aspetti della procedura che riguardano il mantenimento della sterilità fino a quando la confezione sterile non sia aperta o danneggiata. La persona fisica o giuridica redige una dichiarazione in cui afferma che la sterilizzazione è stata eseguita secondo le istruzioni del fabbricante. 4. Se il sistema o kit procedurale contiene dispositivi che non recano la marcatura CE o se la combinazione di dispositivi scelta non è compatibile con la destinazione d'uso originaria, o se la sterilizzazione non è stata eseguita secondo le istruzioni del fabbricante, il sistema o kit procedurale è considerato un dispositivo a sé stante ed è soggetto alla pertinente procedura di valutazione della conformità di cui all'articolo 52. La persona fisica o giuridica assume gli obblighi dei fabbricanti. 5. I sistemi o kit procedurali di cui al paragrafo 1 del presente articolo non recano una nuova marcatura CE, bensì il nome, la denominazione commerciale o il marchio registrato della persona di cui ai paragrafi 1 e 3 del presente articolo e l'indirizzo presso il quale tale persona può essere contattata, in modo tale che essa posa essere localizzata. I sistemi o kit procedurali sono corredati delle informazioni di cui all'allegato I, punto 23. La dichiarazione di cui al paragrafo 2 del presente articolo è tenuta a disposizione delle autorità competenti, dopo l'assemblaggio del sistema o kit procedurale, per il periodo applicabile ai dispositivi combinati conformemente all'articolo 10, paragrafo 8. In caso di periodi di durata diversa, si applica quello di maggior durata.
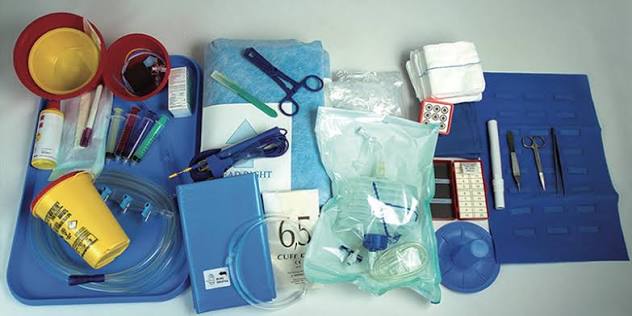
Fig. 1 - Set chirurgico (Art. 22 MDR) segue allegato Certifico Srl - IT | Rev. 0.0 2026 MDR Regolamento dispositivi medici | Reg. (UE) 2017/745
|
||
 |
||
|
sono siti di: Se vuoi cancellarti dall'invio della newsletter clicca qui oppure effettua il login al sito ed entra nella Tua Area Riservata, in “Modifica dati” agisci con la spunta sul box di selezione “newsletter”. L'elenco completo di tutte le ns newsletter è qui: Archivio newsletter certifico.com
Testata editoriale iscritta al n. 22/2024 registro periodici Tribunale di Perugia 19.11.2024 |
||
| Certifico Srl 2000-2026 | VAT IT02442650541 | ||


Certifico s.r.l.
Sede: Via A. De Curtis, 28 - 06135 Perugia - IT
Sede: Via Madonna Alta 138/A - 06128 Perugia - IT
P. IVA: IT02442650541
Tel. 1: +39 075 599 73 63
Tel. 2: +39 075 599 73 43
Assistenza: 800 14 47 46
www.certifico.com
info@certifico.com
Sede: Via Madonna Alta 138/A - 06128 Perugia - IT
P. IVA: IT02442650541
Tel. 1: +39 075 599 73 63
Tel. 2: +39 075 599 73 43
Assistenza: 800 14 47 46
www.certifico.com
info@certifico.com
Testata editoriale iscritta al n. 22/2024 del registro periodici della cancelleria del Tribunale di Perugia in data 19.11.2024